肝豆状核变性是以青少年为主的遗传性疾病,由铜代谢障碍引起。其特点为肝硬化、大脑基底节软化和变性、角膜色素环(Kayser-Fleischer环),伴有血浆铜蓝蛋白缺少和氨基酸尿症。又名Wilson病。
-
挂什么科:内科 神经内科
-
需做检查:脑脊液铜 尿铜 尿铜(Cu) 尿铜蓝蛋白 大生化检查 乳酸与丙酮酸比值 血浆铜蓝蛋白(CP) 血清铜(Cu2+ 颅脑超声检查 脑电图检查 语调与语态 颅脑CT检查 血清铜 肾功能检查
-
治疗方法:药物治疗 手术治疗 中医治疗 对症治疗
-
常用药物:暂无相关信息
-
一般费用:根据不同医院,收费标准不一致,市三甲医院约(5000-20000元)
-
传染性:无传染性
-
治愈周期:1-3个月
-
治愈率:20%
-
患病比例:0.0002%
-
好发人群:好发于10~25岁的青少年
-
相关症状:
-
相关疾病:
虽在婴儿期肝脏就已有铜的蓄积,但6岁前罕有肝病症状发生,而50%在15岁前发病,偶有60岁才发病者,初起症状42%为肝病表现,34%为神经系统,10%为精神症状,12%为继发于肝病的内分泌或血液系症状,1%为肾损害表现,约25%患者同时出现两个以上系统受累表现。
一、肝脏
以肝病为初起发病者年龄往往较小。其临床表现差异很大,可表现为急性或慢性肝炎、暴发性肝功能衰竭或肝硬化,因而,Wilson病肝脏病变所致临床表现没有特异性。在无症状期或肝硬化早期,肝功能可正常,或仅有轻微的转氨酶增高,多起病隐匿,呈现一慢性病程。开始有乏力、疲劳、厌食、黄疸、蜘蛛痣、脾肿大和脾功能亢进,最终导致门脉高压、腹水、曲张静脉出血以及肝功能衰竭。
Wilson病患者肝脏常缩小或正常大小,有坏死后肝硬化特点,可以腹水、食管静脉曲张破裂出血为初发表现。另外临床表现、生化检查、组织学检查酷似慢性活动性肝炎者也较多见。有些患者偶尔发现K-H环或出现神经精神症状后才想到本病而得到诊断。所以,对35岁以下,HBsAg阴性的慢性肝病患者,应想到本病并做化验检查以确立诊断。
二、神经系统
神经系统表现一般出现在12~30岁患者,几乎都同时伴有K-F环。开始起病轻,但如得不到及时治疗则很快向严重程度发展。早期有腕部震颤、扮鬼脸、口吃和书写困难等,同时有步态僵直、吞咽困难,四肢呈波动性强直,表情贫乏和固定,不断流涎,智力尚可。脑电图为非特异性慢波,无助于诊断。另外,此时CT检查,脑诱发电位都无特异性表现。MRI对衡量大脑、小脑及脑干病变较CT敏感,但无症状者常正常,肝功能检查多正常。
三、精神症状
表现为行为异常,躁狂抑郁或精神分裂症、痴呆。至少有四方面精神障碍:情感异常、行为异常、精神分裂症和认知障碍。有以上几方面表现时,治疗往往只能部分得到缓解。
四、眼
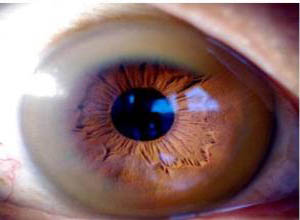
K-F环位于角膜周围缘的膜后弹力层,呈棕色或绿色,或金黄色,宽可达2mm,用斜照灯或肉眼即可看到。此色素环与铜颗粒的分布、密度及大小有关。K-F环几乎总与神经系统症状相伴行,但在无症状儿童或肝脏受损,特别是伴慢性活动性肝炎者,可无K-F环。K-F环的出现有助于诊断,但并不是Wilson病的特征性表现。如儿童时期长期肝内胆汁淤积、慢性活动性肝炎伴肝硬化和隐原性肝硬化都会由于胆汁排泄铜障碍而导致角膜及其他器官铜淤积。
葵花状白内障也是Wilson病少见的一个眼部表现,常与K-F环并存,此特征在应用D-青霉胺治疗时常较K-F环消失快。
五、血液系统
Wilson病程中常出现急性血管内溶血,至少15%的患者溶血表现明显。溶血常是短暂性的和自限性的,常较肝病表现超前数年,溶血发生时常无K-F环发生。因而,对于20岁以下的溶血患者,应从生化检查上除外其他原因导致的溶血,Wilson病溶血患者,血Coombs试验阴性,属非球形红细胞性溶血。偶有急性溶血与急性肝功能衰竭同时出现者,预示病情重,常在数周内死于肝或肾功能衰竭。
导致溶血的原因不明,有人认为是由于肝脏在短期内大量释放铜入血,红细胞大量摄取铜,导致对细胞膜和血红蛋白的氧化损伤,也有人认为铜的毒性作用是对细胞膜磷脂的氧化作用。
另外,以肝脏损伤为主的患者,急性肝功能衰竭,严重失代偿性肝硬化,凝血因子合成减少、血小板质量差,脾大可致血小板减少和白细胞减少,这些合Wilson病患者存有出血倾向。
六、肾脏
Wilson病肾功能受损程度不一,包括肾小球滤过率降低,肾血流量减少和肾小管病变。其中近曲小管受累可有氨基酸尿、糖尿、尿酸增高(伴血清尿酸低)、高尿磷、高尿钙、蛋白尿,后者包括低分子球蛋白和胶原分解产生的羟脯氨酸多肽;远曲小管受累pH降至5.2以下,这也是肾结石形成的原因,青霉胺可明显改善肾功能,但偶见青霉胺致肾病综合征和Goodpastures样综合征样的严重副作用。
七、骨骼
可有脱钙,骨质软化,佝偻病,自发性骨折,关节下囊肿,骨关节痛,分离性骨软骨炎和软化钙化症,临床症状常不明显,患者可有膝关节或其他大关节疼痛和僵硬。
八、其他
心脏可有心律失常,心肌病和植物神经功能异常,继发于肝病的内分泌变化,年青女性有闭经,男性发育迟缓,乳房发育,胰腺受损有胰功能不全和糖尿病,指甲弧呈蓝色,含铜量增加。
关于本病自然病程 理论上分四期:Ⅰ期:铜在肝细胞质原始聚积直至达饱和状态,临床上无症状;Ⅱ期:铜从胞质转入溶酶体,部分释放入血。大多数(60%)患者铜的再分布是逐渐发生的,临床表现不明显,但是,如此过程进展快,血铜突然升高可致溶血,肝内快速再分布可致肝坏死或慢性活动性肝炎,可能发生肝功能衰竭;Ⅲ期:肝外组织铜贮积,出现肝硬化、神经、角膜和肾损害,临床有相应表现,可出现溶血,可死于肝功能衰竭,也可以再度缓解为无症状;本期表现多样,如肝硬化进展慢、肝外铜贮积慢,患者可多年无症状,但进展快则临床经过凶险;Ⅳ期:即络合物长期治疗后的缓解期。
病因学
本病的基本病因是体内各组织,尤其是肝、脑、肾、角膜等处铜沉积过多,导致组织损害和病变。
发病机理
关于肝豆状核变性发生机制有以下几个学说:①该类患者胆汁中与铜结合的正常物质缺陷,可能是鹅脱氧胆酸与牛磺酸结合缺陷,导致胆汁分泌铜功能障碍。不支持这一学说的证据是:该类患者胆汁铜结合蛋白没有质的改变,而且没有证据表明该类患者存在胆酸代谢异常。②肝脏铜结合蛋白合成异常,导致蛋白对铜的亲合力增加,支持这一学说的证据有,Wilson病患者铜结合蛋白(肝脏铜蛋白)对铜的结合常数是原发性胆汁性硬化患者该常数的4倍,但人们对资料的分析方法提出质疑,因而,Wilson病中异常蛋白对铜的高亲和力是否就是Wilson病的形成机制有待进一步阐明。③最合理的学说,是肝脏细胞的溶酶体参与了铜的代谢过程,实验观察到,Wilson病患者肝细胞溶酶体含量高出对照的40倍,认为Wilson病患者肝细胞溶酶体缺陷干扰了铜由溶酶体分泌到胆汁中去的过程,从而导致了Wilson病患者肝脏含铜量的增加。
总之,Wilson病不是由于肠道对铜吸收增加,而是由于胆道对铜的分泌障碍,这一障碍是先天性的,有基因缺陷的患者,在生后3个月不能合铜正平衡代谢转为正常,使铜正平衡代谢持续存在,结果导致了铜在体内的堆积。
病理改变
一、肝脏
最早的组织学变化是光镜下小叶周边区的肝细胞核内糖原变性。核内糖原成块或呈空泡状,有中等程度的脂肪浸润。脂肪滴由甘油三酯组成,以后脂肪滴数量增加,并融合增大。脂肪变性从形态学上与酒精脂肪变性相似,与脂肪变性并存或在脂肪变性前就已发生病变的细胞器是线粒体,线粒体体积增大、膜分离、嵴扩张、呈晶体状排列,有空泡、基质呈显著颗粒状。线粒体变化可能在发病机制上与脂肪变性有关。随着D-青霉胺的治疗,线粒体变化可以减轻甚至消失,说明线粒体的变化是由铜的毒性作用引起。
肝脏由脂肪浸润到肝硬化这一过程的变化速度个体差异很大。某些患者可发生慢性活动性肝炎,可有单核细胞浸润,多数为淋巴细胞和浆细胞,可有碎屑样坏死,并且这种坏死可越过界板,可有肝实质塌陷,桥接样坏死和肝纤维化,这种慢性活动性肝炎相鉴别。肝脏病变可自然缓解,可进展为大结节性肝硬化或很快发展为暴发性肝炎,后者治疗效果很差。
在肝硬化形成过程中可能有肝实质的炎性细胞浸润或实质坏死,最终形成大结节性或大结节小结节并存的肝硬化,纤维分隔可宽可窄,胆管增生,还可伴有Wilson病早期的一些病理变化如核糖体变性、脂肪变性等。
二、脑
整个神经系统均可受累,而以豆状核、视丘、尾核、脑岛和带状核受累较重,以豆状核内的壳核最为显著。肉眼可见大脑半球有不同程度萎缩,豆状核缩小、软化和小空洞形成,组织学显示神经细胞变性坏死,星形胶质细胞肥大、增生和变性。
三、肾脏
铜在近曲小管沉着,显示脂肪变性和水样变性。
四、角膜
铜沉着在角膜后Descemet膜的周围形成棕绿色的色素沉着,称Kayser-Fleischer环。
诊断
1.凡具下列情况应高度怀疑HLD患者,都必须行裂隙灯检查有无角膜K-F环和铜代谢测检。
(1)已证实HLD患者的同胞。
(2)同胞中有幼年死于急性重型肝炎(暴发性肝炎)或其他肝病(尤其病毒性肝炎血清抗原抗体阴性)者。
(3)儿童或少年发生原因不明的肝硬化,一过性黄疸,流涎,震颤,舞蹈样运动或精神错乱,均需注意与HLD鉴别,必要时,需进一步行裂隙灯和铜代谢检查。
2.诊断标准
(1)家族遗传史,父母是近亲婚配,同胞有HLD患者或死于原因不明的肝病者。
(2)缓慢进行性震颤,肌僵直,构语障碍等锥体外系症状,体征及(或)肝症状。
(3)肉眼或裂隙灯证实有K-F环。
(4)血清铜蓝蛋白<200mg/L。
(5)尿铜>50μg/24h。
(6)肝铜>250μg/g(干重)。
判断:凡完全具备上述(1)~(3)项或(2)及(4)项者,可确诊为临床显性型,仅具有上述(3)~(5)项或(3)~(4)项者属无症状型HLD,仅有(1),(2)项或(1),(3)项者,应怀疑HLD。
鉴别诊断
本病临床表现复杂,患者无神经系统表现,出现各系统症状时临床误诊相当普遍,鉴别应从肝脏及神经系统两方面考虑。
1.Mekes病及慢性肝病由于蛋白严重缺乏,血清CP可下降,胆汁性肝硬化也可出现K-F环,须注意鉴别;
2.本病出现帕金森病某些体征,可根据角膜K-F环,严重共济失调性震颤,血清铜蓝蛋白降低等与PD鉴别;
3.还须与急性或慢性肝炎,肝硬化,小舞蹈病,Huntington舞蹈病,扭转痉挛,老年性痴呆,精神病,肝肾综合征等鉴别。
肝豆状核变性西医治疗
1、本病一经诊断或患者出现神经系统体征前就应进行系统治疗,有效防止病情发展,治疗愈早愈好,注意减少食物含铜量(<1mg/d),限制含铜多的饮食,如坚果类,巧克力,豌豆,蚕豆,玉米,香菇,贝壳和螺类,蜜糖,动物肝和血等,高氨基酸,高蛋白饮食能促进尿铜排泄。
2、药物治疗
(1)、青霉胺:为首选药,成功的关键是早期诊断早期治疗。初始剂量为每日1~2g,分4次餐前服用,可逆转或减轻肝、神经和精神病变。病变缓解快慢个体差异很大,可数星期迅速康复,也可数年不见好转,有时甚至出现神经症状加重,后者可加大青霉胺用量至每日4g,数年后,症状明显改善,病情稳定后可减至每日1g,终身服药。副反应有过敏反应,白细胞和血小板减少,再生障碍性贫血,蛋白尿和红斑狼疮样综合征。如遇过敏反应可脱敏后再用。
(2)锌制剂:可抑制铜在肠道内的吸收,锌可促使肠道产生铜结合蛋白,使铜与肠粘膜隔离,用硫酸锌或醋酸锌制剂每次50mg,每日3次,餐间服。
3、对症治疗
肌强直和震颤可用安坦或金刚烷胺,症状明显可用复方L-dopa;精神症状明显可用抗精神病药,抑郁症状明显可用抗抑郁药,智力减退可用促智药,无论有无肝损害,均需护肝治疗,可用肝泰乐,肌苷及维生素C等。
4、中医治疗
中医认为本病属肝阴不足,肝风内动,可用肝豆片1号及肝豆汤,由大黄,黄连,姜黄,金钱草,泽泻和三七等组成,中药能促进胆汁,尿及粪的排铜,较安全,但排铜作用不如青霉胺及锌剂等,单独使用常不能达到满意疗效。
5、手术治疗
①WD合并脾功能亢进,白细胞及血小板长期显著减少,易发生出血和感染,脾切除是重要辅助治疗措施,青霉胺也有降低白细胞及血小板副作用,这类患者宜小剂量或不用青霉胺;②治疗无效严重病例可肝移植治疗,文献报告2例WD患者肝移植后,症状改善,K-F环消失,生化指标恢复正常,术后分别存活6年和4年,目前国内WD患者肝移植最长存活时间为500天。
6、基因治疗 目前仍处于实验性研究阶段。
预后
本病如不及时进行积极治疗,病情多数持续进行,至晚期则因严重肝硬化,肝功能衰竭或并发感染而死亡,病程长短与起病年龄有密切关系,进展多数缓慢,病程可延续数年甚至三四十年,平均病程为4~5年,但持续十多年者并非少见,在1948年以前,本病因无有效疗法,病程多不超过3~4年,自从应用化学疗法以来,预后颇有改观,经化学疗法后,神经症状可有一定程度的好转,但亦可复发,有些症状可完全缓解一段时间,治疗以前神经症状存在越久者,恢复的程度就越差,有些神经症状,在治疗连续进行2年以上仍可继续好转,但肝脏损害恢复较差,本病的预后主要取决于治疗的早晚,发现本病时肝脏的情况以及肝脏疾病进展的快慢,有些患者可早期死于急性肝坏死,亦有人认为神经症状出现越早者病情进展越快,另有人提出少年型的药物疗效比成年人差,曾报告一组10例少年型病例,平均病程为5~7年,本病如能及早治疗,症状(特别是神经症状)可能停止发展或获得缓解,因此对于患者的同胞兄弟在出现症状之前,应从早进行检查,治疗越早,预后将越佳,今后对于代谢障碍各个环节如能更进一步了解,则疗效必将进一步提高。
肝豆状核西医治疗
当前疾病暂无相关疗法。 (仅供参考,详细请询问医生)
不适宜食物:含铜高的食物如:动物肝、动物血、猪肉,蛤蜊、牡蛎、田螺等贝壳类,坚果类,黄豆、黑豆、小豆、扁豆、绿豆等干豆类,芝麻、可可、巧克力、明胶、樱桃等,蘑菇、荠菜、菠菜、油菜、芥菜、茴香、芋头、龙须菜等;对铜制餐具、食具也应慎用;兴奋神经系统的食物,如浓茶、咖啡、肉汤、鸡汤等食物,适宜食物:低铜食物:如精白米、面、瘦猪肉、瘦鸡鸭肉、马铃薯、小白菜、萝卜、藕、茎蓝、桔子、苹果、桃子及砂糖、牛奶(不仅低铜,而且长期服有排铜效果)。去油骨头汤、蛋黄等含钙丰富的食物。
食疗方
1、玉米30g,加水适量煮服,玉米汤代茶饮用。
2、高粱米30g,羊肉30g,葱盐适量。加水同煮成羊肉高粱米粥饮用。
3、鳗鲡鱼,切段。加葱,盐炖服。
4、红烧鳝鱼。切段,加酱油及适量白糖,煮食。
(以上资料仅供参考,详细情况询问医生)
1、防治本病应及早确诊,及时纠正患者铜代谢的正平衡状况,注意减少食物含铜量(<1mg/d),限制含铜多的饮食,如坚果类,巧克力,豌豆,蚕豆,玉米,香菇,贝壳和螺类,蜜糖,动物肝和血等,高氨基酸,高蛋白饮食能促进尿铜排泄。
2、对WD患者的家族成员测定血清铜蓝蛋白,血清铜,尿铜及体外培养皮肤成纤维细胞的含铜量有助于发现WD症状前纯合子及杂合子,发现症状前纯合子可以及早治疗,杂合子应禁忌与杂合子结婚以免其子代发生纯合子,产前检查如发现为纯合子,应终止妊娠,以杜绝患者的来源。
实验室检查
1.血清CP及血清铜氧化酶活性测定是本病重要的诊断依据
①血清铜蓝蛋白测定:WD患者血清CP<0.2g/L(正常值0.26~0.36g/L),甚至为零,血清CP值与病情,病程及驱铜疗效无关,不能作为病情监测或疗效观察指标;新生儿血清CP值只有正常人的1/5,以后迅速升高,2~3个月达到成人水平;12岁前儿童血清CP矫正公式为:矫正后CP值=血清CP测定值×[(12-年龄)×1.7];须注意血清CP降低还见于肾病综合征,慢性活动性肝炎,原发性胆汁性肝硬化,某些吸收不良综合征,蛋白-热量不足性营养不良等;
②WD患者血清CP氧化酶活力<0.2密度(正常值0.2~0.532光密度)。
2.微量铜测定
①血清铜测定:正常人为14.7~20.5mmol/L,90%的WD患者血清铜降低,血清铜与病情和疗效无关,原发性胆汁性肝硬化,慢性活动性肝炎,肾病综合征及严重营养不良等患者血清铜也可降低;
②尿铜测定:多数WD患者24小时尿铜量显著增加,服排铜药后尿铜进一步增高,体内蓄积铜大量排出后尿铜量渐降低,尿铜量可作为临床调整排铜药剂量参考指标;WD患者通常尿铜量>200μg/24h(正常<50μg/24h),个别高达1200μg/24h,少数患者正常或稍高;青霉胺负荷试验:口服青霉胺后正常人和未经治疗患者尿铜明显增高,但患者更显著;慢性活动性肝炎,原发性肝硬化等尿铜量也增高;
③肝铜量测定:是诊断WD金标准,由于难以普遍接受肝穿刺,不能作为常规检查,生化检查不能确诊的病例测定肝铜量是必要的,正常肝铜含量50μg/g干重,WD患者多为250μg以上/g干重,杂合子及肝病患者肝铜含量虽可增高,但不超过250μg/g干重,穿刺肝组织恰为新生肝硬化结节可出现假阴性;
④离体培养皮肤成纤维细胞铜含量测定:最早由Chan等(1980)报道,国内陈嵘等(1994)建立稳定的离体培养皮肤成纤维细胞模型,对WD患者,杂合子及正常人皮肤成纤维细胞离体传代培养,经高浓度铜孵育后WD患者胞浆内铜/蛋白比值远高于杂合子组及正常对照组,二者完全无重叠,可确诊不典型病例;
⑤放射性铜测定:口服或静脉注射64Cu或67Cu后,示踪观察它与铜蓝蛋白动力学变化,健康人放射性铜进入血液与血浆蛋白结合,出现第一次血浓度高峰,放射性铜进入肝脏并与肝铜蛋白(包括Apo-CP)结合,血浆放射性铜浓度下降,带放射性铜的肝铜蛋白释放入血出现第二次血浓度高峰;患者可出现四种异常:肝脏摄取铜障碍使第一次放射性血铜浓度高峰延长;放射性铜与CP结合障碍不出现第二次浓度高峰;胆道排铜障碍使粪便中放射性铜排泄减少而尿中排泄增加;放射性铜在体内转换延长。
3.肝肾功能检查 某些WD患者早期可无肝功能异常,肝损害可出现不同程度肝功能异常如血清总蛋白降低,γ-球蛋白增高等;肾功能损害可出现血清尿素氮及肌酐增高,尿蛋白等。
影像学检查
1.骨关节X线检查 约96%患者骨关节X线异常,双腕关节最常受损,表现骨质疏松,骨关节炎,骨软化,关节周围或关节内钙化,自发性骨折和脊椎骨软骨炎等。
2.神经影像学检查 CT异常率约85%,CT显示双侧豆状核对称性低密度区有诊断价值,常见侧脑室和第Ⅲ脑室轻度扩大,大脑和小脑沟回变宽,脑干萎缩,红核及齿状核低密度,治疗后影像学无改变,MRI可见双侧豆状核对称性受累,T2W呈同心板层型增强,黑质致密带,大脑导水管周围灰质及大脑脚高信号,丘脑较少受累。
3.脑电图检查 约50%的WD患者出现异常,EEG改变多与病变严重程度一致,青霉胺及二巯基丙醇治疗后EEG可改善。
4.诱发电位检查 可证实本病感觉系统亚临床损害,脑干听觉诱发电位(BAEP)异常率最高,各波潜伏期和波峰间期延长;视觉诱发电位(VEP)表现N1,N2,P1波PL延长;体感诱发电位(SEP)也有改变。
5.正电子发射断层扫描(PET) WD患者可显示脑局部葡萄糖代谢率(rCMRG)降低,豆状核明显,rCMRG改变可早于CT改变,对WD早期诊断颇有价值。
6.基因诊断 WD患者及家系成员常规生化检测发现,在患者,杂合子与正常人间存在10%~25%的重叠数据,影响检测特异性,基因诊断对症状前诊断及杂合子检出具有优越性,
①限制性片段长度多态性(RFLP)连锁分析:Figus等(1989)首次应用RFLP对17个家系未患病个体进行基因检测,国内外学者用此法对许多WD家系进行连锁分析,检出不少症状前患者及表型正常杂合子或病理基因携带者;
②微卫星标记分析:1993年国外已克隆了WD的cDNA片段,获得WD基因附近几个微卫星标记,Thomas用几个新的微卫星DNA对WD患者家系分析,提出单倍型有助于WD家系鉴定及其他成员诊断;
③半巢式PCR-酶切分析:直接检测WD患者14号染色体外显子His1069GLn基因突变;
④MspI酶切法:马少春等(1998)研究发现,中国WD患者中8号外显子上778密码子突变者占28.8%,是中国WD患者高频突变位点;
⑤荧光PCR法:黄帆等(1999)用荧光PCR技术,在58个WD家系66例患者中诊断5例有Arg778Leu突变的纯合子,21例杂合子,总检出率为39.4%,较酶切法敏感。
肝豆状核变性患者免疫功能部分低下,部分患者有假性延髓麻痹的症状如吞咽困难,饮水返呛等,特别是长期卧床的病人更容易患坠积性肺炎,尿路感染与褥疮,有锥体外系症状的患者,行走困难,易跌倒而出现骨折,肝豆状核变性患者在肝硬化失代偿期有门静脉高压合并食管胃低静脉曲张者,易出现急性上消化道出血,甚至发生出血性休克;少数肝脏的解毒能力下降,易出现肝性脑病,肝肾综合征等;亦有患者由于脑部损害而合并癫痫发作,上述种种并发症往往加重病情,严重影响了治疗效果,使患者住院时间延长,如不及时,准确的处理,部分患者预后较无并发症的患者差。