郎格罕细胞性组织细胞增生症(Langerhans cell histiocytosis,LCH)为组织细胞增生症Ⅰ型。郎格罕细胞组织细胞增生症是一种单克隆起源的树突状细胞增生性疾病,临床表现多样,病变可为局灶性或播散性,是一组发病缓急、临床症状和病变范围差异很大的综合征,迄今为止,病因尚未明了。随着化疗方法的进步,近年来此病的预后得到了明显的改善。
-
挂什么科:儿科 小儿内科
-
需做检查:染色体 胸部平片 骨髓增生程度 浆细胞 血常规 细胞组织化学染色 肾功能检查 四肢的骨和关节平片 涂片
-
治疗方法:药物治疗 放射治疗 支持性治疗
-
常用药物:暂无相关信息
-
一般费用:根据不同医院,收费标准不一致,市三甲医院约(5000-10000元)
-
传染性:无传染性
-
治愈周期:3-6个月
-
治愈率:50%
-
患病比例:0.0001%
-
好发人群:儿童
-
相关症状:
-
相关疾病:
临床表现差异大,可呈局灶性或全身性变化,起病可急可缓,病程可短至数周或长达数年,各亚型有相对特殊的临床表现,但可出现过度型或重叠性表现。
1.勒-雷综合征(急性婴儿型) 以内脏病变为主,此型临床表现最严重,也最多见,多于1岁以内发病,起病急而重,以内脏和皮肤受侵害为主,表现为发热,皮疹,咳嗽,苍白,耳流脓,肝,脾很快增大,肺部广泛浸润,淋巴结轻度肿大,热型不规则,以周期性或持续性高热多见,偶见稽留热,皮疹较特殊,主要分布于躯干,颈部,头皮和发际,四肢少见,初起为淡红色斑丘疹,直径1~3mm,继而呈出血性或湿疹样,皮脂溢出样皮疹,以后皮疹表面结痂,脱屑,脱痂后留有白斑或色素沉着,各期皮疹可同时存在,常成批发生,一批消退,一批又起,手触摸时有粗糙感,出疹前常先发热,出疹同时伴肝,脾增大,疹退热降,肝,脾亦缩小,常有轻咳,伴有呼吸道感染时,症状急剧加重,极易发生肺炎,出现喘憋和发绀,但肺部体征多不明显,因系肺间质性病变,可并发气胸和皮下气肿,呼吸衰竭是致死的主要原因,此外常见耳流脓,营养不良,腹泻和贫血,也可同时有溶骨性骨骼病变,但与其他2型相比,相对较少,若不治疗常于6个月内死亡。
2.韩-薛-柯综合征 骨损害伴中度其他器官侵犯,又称慢性黄色瘤病,多发生于3~4岁小儿,颅骨缺损,突眼,尿崩症是此型的三大特征,这三大特征可先后出现或在病程中只见其中之一或二,初起颅骨损害呈肿块状凸起,硬,有轻度压痛,当病变蚀穿颅骨外板后,肿物变软,触之有波动感,常可触及颅骨边缘,压痛不明显,以后肿块逐渐吸收,局部下凹,缺损大者可触及脑,并随脉搏跳动,眼球突出多为单侧,为眶骨破坏所致,尿崩症由垂体或下丘脑受累所致,不一定有蝶鞍破坏,皮疹呈孤立,稀疏的黄色丘疹,似半个小米粒或黄豆状,突出在皮肤表面,若以玻片压于其上,可见皮疹中央发黄,是慢性黄色瘤病名称的来由,其后皮疹开始消退变软,变浅,慢慢吸收,此外低热,轻度肝,脾增大,贫血,牙龈齿槽肿胀,发炎,坏死,萎缩,牙齿松动,脱落也常见,肺部受累重者可见气喘和发绀,本型病程迁延,病变新旧交替,最终多数病人恢复健康。
3.骨嗜酸细胞肉芽肿 为单纯骨损害型,是本症中预后最好的一型,多发于4~7岁小儿,但也可见于婴儿或成人,任何骨骼均可受累,但以颅骨,四肢骨,脊椎,骨盆最常见,病灶多为单发,亦可多发,患者除骨骼病变外多无其他症状或仅有低热,不少患儿是在偶然情况下或出现病理性骨折时发现,唯有脊椎病变的患儿,特别是发生椎弓破坏者,常伴神经压迫症状,如肢体麻木,疼痛,无力,瘫痪,甚至大小便失禁成为疾病的主诉而就医。
(一)发病原因
LCH的病因及发病机制至今尚不明了,可能的病因学包括感染,免疫功能紊乱和肿瘤等学说,一般认为,LCH无遗传倾向,目前多认为本病是一种免疫性疾病,关于LCH病变的性质仍是争论的热点问题,尽管有少数病例最终发展为恶性肿瘤,但多数研究认为它是一种免疫性疾病,目前尚无研究证实细菌,真菌感染与LCH的相关性,病毒曾经被疑为LCH的病因,有人提出LCH可能是感染人类疱疹病毒-6(HHV-6)和人类巨细胞病毒(HCMV)所致,但至今尚缺乏有力证据,HHV-8感染在LCH发病中所占的位置也尚未被确定,总之LCH的超微结构研究未发现病毒颗粒或病毒特异细胞产物。
(二)发病机制
1.发病机制 正常的LC起源于骨髓的CD34 干细胞,是一种抗原递呈细胞(antigen-presenting-cell,APC),可分泌多种具有生物活性的细胞因子,pLCs产生IL-1α,IL-1β,IL-4,TNF-α,INF-γ,GM-CSF,TGF-α和TGF-β等多种细胞因子,这种细胞因子“风暴”是LCH发病机制的重要组成部分并与LCH的临床特征,如炎性增生,坏死,纤维化和溶骨性病变相符,巨噬细胞,嗜酸性粒细胞,巨细胞(giant cell),淋巴细胞,特别是淋巴细胞产生的细胞因子在LCH病变中也起到非常重要的作用, LCH的增殖程度接近炎症性组织而低于肿瘤性组织,有学者认为pLCs克隆性增生是对炎症刺激的反应,类似于其他炎症性疾病时T,B或NK细胞的克隆性扩增,病变为良性的,可以修复;而病变是恶性的则可以扩散并有导致致命性结果的高危因素,这可以解释其临床上的症状的多变性,也可以解释成人肺的LCH只是肺部的pLCs的局部浸润,关于LCH是炎性反应还是肿瘤性增生观点不一,但可反映出一些对感染反应性增殖的病例能够发展成为肿瘤性疾病这一事实。
例如人类疱疹病毒8(HHV8)感染所致的卡波肉瘤(Kaposis sarcoma)就是代表之一,Yousem等对成人肺LCH的克隆性进行分子水平的分析发现,只有29%的病变组织显示克隆性,因此不支持肺的LCH是肿瘤性的疾病的假设,肉芽肿被认为是阻止细胞内病原体扩散的生理反应,在肉芽肿时这种细胞增殖与凋亡之间的平衡对高度的细胞逆转和网状聚集起着至关重要的作用,多种原因不明的炎性疾病如良性淋巴肉芽肿病(sarcoidosis),克罗恩病(Crohns病)和韦格纳肉芽肿病(Wegeners granulomatosis)均有肉芽肿,这种改变对组织起损伤作用而非保护作用, Geissmann等人的研究证明,在疾病的不同过程中,pLCs的分化程度不同,慢性或骨浸润患者的pLCs表达不成熟的树突细胞表型(如CD68,CD14),而不表达细胞膜主要组织相容复合物(MHC)Ⅱ类分子或共刺激分子CD86,同时IL-10为阳性,在成人肺部LCH患者,pLCs表达CD86,IL-10阴性,这些结果进一步提示成人肺LHC与慢性或骨浸润LHC可能是临床及病理生理过程不同的疾病,前者为炎症后纤维增生而非肿瘤过程,同时也可以推测由CD1-的巨噬细胞产生的IL-10在维持pLCs不成熟的特性上起着至关重要的作用。
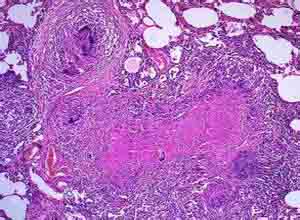
2.病理学改变 LCH的病理学改变为增生性肉芽肿,pLCs光镜下为单个核细胞,直径约13µm,胞浆为均匀的粉红色,核弯曲呈咖啡豆样或呈多叶状,核染色质不规则,含有1~3个嗜碱性的核仁,核仁明显,电镜下,细胞质边缘不规则,伪足易见,胞浆量丰富,含有数量不等的散射状细胞器,如粗面内质网,游离多核糖体,溶酶体和线粒体等,pLCs的胞浆内含有一种LC所特有的特殊的细胞器-郎格罕细胞颗粒或称Birbeck颗粒,这种颗粒呈长板状,长度190~360nm,宽度为33nm,中央有纹状体,有时末端可见囊状扩张,呈网球拍状或棒状,病灶部位除可见LC外,尚可见到嗜酸性粒细胞,巨噬细胞和淋巴细胞不同程度的增生。
此外还有合体多核巨细胞,少数中性粒细胞及浆细胞,随着病情的进展,病变部位可纤维化或黄色瘤样改变,并可见局灶性出血,坏死及含有血黄素颗粒的巨噬细胞,同一器官中可同时出现增生,坏死或纤维化等不同阶段的改变,病变早期仅是单一非充脂LC,病变越久,充脂性细胞(泡沫细胞)越易见,病程长者则可见大量充脂LC和嗜酸性粒细胞,或以嗜酸性粒细胞为主形成肉芽肿,增生中心常有坏死,通过对LCH标本的冷冻石蜡包埋,经免疫过氧化物酶染色的病理学研究发现郎格罕细胞及pLCs细胞表达MHC的Ⅰ类和Ⅱ类蛋白,CD1a,CD4,CD14,S-100,CD74和波形蛋白(vimentin),同时还表达CD25,IFN-γ,CD11b,CD35,CD21和fascin以及黏附分子CD54和CD58,这些分子可以在激活的郎格罕细胞中检测到, 郎格罕细胞经ATP酶,α-甘露糖和S-100蛋白免疫组织化学染色均呈阳性反应,这些免疫组织化学染色已作为LCH的诊断依据之一。
诊断
需临床,放射线及病理三方面配合诊断,发热,贫血,肝脾肿大,耳流脓伴典型皮疹时,要考虑郎格罕细胞性组织细胞增生症Ⅰ型,突眼,尿崩症,颅骨缺损是Ⅱ型综合征典型表现,应疑及本病,典型的骨X线变化,皮疹印片或活检,骨病变处的病理检查见到典型的郎格罕细胞是诊断的依据,实验室检查S-100蛋白染色阳性,电镜找到Birbeck颗粒支持本病的诊断, 1987年组织细胞协会写作组提出了诊断LCH可信的三级标准,这一标准已被国际上广泛采用,它有助于LCH与其他组织细胞增生症的鉴别,也有利国际间的交流。
鉴别诊断
1.本病应与以下组织细胞增生症相鉴别
(1)窦性组织细胞增生症伴块状淋巴结肿大(sinus histiocytosis with massive lymphadenopathy,SHML):常表现为双侧颈淋巴结无痛性肿大,其他部位肿大淋巴结及结外病变如皮肤,软组织和骨损害可见于近半数患者,皮肤病变为黄色或黄色瘤样,骨病变为溶骨性损害,因此临床表现及X线表现均与LCH难以鉴别,SHML的组织学改变为组织细胞呈窦性增生,常与其他淋巴样细胞和浆细胞相混,病变细胞无LC细胞典型的核凹陷特点,CD1a抗原阴性,电镜下无Birbeck颗粒。
(2)嗜血细胞性淋巴组织细胞增生症:嗜血细胞性淋巴组织细胞增生症(hemophagocytic lymphohistiocytosis,HLH)包括家族性嗜血细胞性淋巴组织细胞增生症(familial hemophagocytic lymphohistiocytosis,FHL)和病毒相关性嗜血细胞综合征(viral associated hemophagocytic syndrome,VAHS)又称继发性嗜血细胞综合征, HLH属常染色体隐性遗传病,是一组以发热,全血细胞减少及肝,脾肿大为特征的临床综合征,自然病程短,预后差,在FHL发现约10%的病例有染色体9q21.3-22异常,20%~30%的病例有染色体10q21-22异常,发病者多为6个月以内婴儿,近50%的病例有阳性家族史,近30%患儿父母有近亲婚配史,高三酰甘油血症(>2.0nmol/L),低纤维蛋白原血症和脑脊液中淋巴细胞增多为FHL的典型改变,患儿无皮疹,无骨骼浸润,病灶中无嗜酸细胞浸润可与LCH鉴别,但若缺乏阳性家族史,临床上与VAHS难以鉴别,因此在诊断时应予以注意, VAHS的临床特点为全血细胞减少,骨髓中单核巨噬细胞增多,并有吞噬血细胞的现象,患儿多有高热,肝脾肿大,肝功能异常和凝血障碍,现认为VAHs除病毒感染外,还可继发于细菌或真菌感染,也称为LAHS(infection associated hemophagocytic syndrome),感染因素去除后可自愈。
(3)恶性组织细胞病(malignant histiocytic disorders):是来源于组织巨噬细胞或组织细胞的少见肿瘤性疾病,其与HLH或SHML等疾病鉴别困难,临床上以成人多见,任何年龄儿童均可发病,常表现为高热,消瘦,衰竭,皮肤黏膜出血,肝脾及淋巴结肿大,脾肿大尤为明显,疾病的早期血象异常不明显,随着病情进展可出现全血细胞减少,病情进展迅速,预后差,病程多不超过6个月,其主要的病理特点为恶性组织细胞或分化较高的组织细胞在肝,脾,淋巴结和骨髓等组织,器官中广泛的灶性浸润,并伴有血细胞被吞噬现象,骨髓涂片中找到典型的异常组织细胞是确诊的重要依据,可与LCH鉴别。
2.与其他疾病鉴别
(1)引起骨病变的疾病:临床上骨髓炎,Ewing肉瘤,成骨肉瘤,骨巨细胞肉瘤等骨肿瘤以及神经母细胞瘤的骨转移均可引起不规则骨破坏,硬化和骨膜反应,在诊断LCH时应注意与这些能引起骨病变的疾病相鉴别。
(2)皮肤病:LCH的皮肤改变多样,无特异性,应与脂溢性皮炎,脓皮病,血小板减少性紫癜等相鉴别。
(3)呼吸系统疾病:LCH的肺部表现常与粟粒性结核相混淆,应注意鉴别。
(4)淋巴网状系统疾病肝,脾及淋巴结肿大者需与霍奇金病,急性白血病,慢性肉芽肿病,尼曼-匹克病等相鉴别。
小儿郎格罕细胞性组织细胞增生症西医治疗
根据病变广泛程度、病情进展速度和发病时的年龄,采取不同的方法。
1、局限病灶手术治疗 病变局限的骨嗜酸性肉芽肿应采取手术刮除或切除。比较小的病灶应用局部氢化可的松注射亦可取得与手术刮除同样的效果。年龄5岁以下尤其3岁以下的易复发或由Ⅰ型发展成Ⅱ型或Ⅲ型,故手术后应进行化疗6个月。年龄大于5岁者术后也应密切观察。
2、放射治疗适用于孤立的骨骼病变,尤以手术刮除有困难的部位如:眼眶周围、颌骨、乳突或负重后易发生骨折和神经损伤的脊椎等部位,以及早期的垂体病变。一般照射量为4~6Gy(400~600cGy)。照射后3~4个月骨骼缺损即可恢复。一般认为尿崩症出现时间较久(如6个月以上),放射治疗大多无效。皮肤病变对放疗亦不敏感。
低危患儿>2岁,伴单一系统疾病,或一个部位骨质或多部位骨质损害,常给予局部治疗,不需全身性治疗。低危患者>2岁伴多系统疾病,但未侵犯血液系统,肝,肺或脾,可给予化疗,常有持续的疗效。
3、药物治疗 肾上腺皮质激素为首选药物,且多药联合治疗并不比采用单一药物效果好,但对多脏器受累病人应采用联合化疗。常用药物:
(1)肾上腺皮质激素:泼尼松(强的松)45~60mg/(m2·d)或地塞米松8~10mg/(m2·d)口服,分3~4次,6周后减至半量,再用4周,然后逐渐减量,总疗程12周。危重病人可静脉滴注氢化可的松250~300mg/m2。急性症状消失后改为口服,此类药物对全身症状如发热、皮疹和贫血等效果较好。
(2)硫酸长春碱:每次6mg/m2,1周1次。或长春新碱每次1.4mg/m2,1周1次,静脉注射,连用4~6周,以后改为每月1次或停8~12周后,再给4~6周,并与泼尼松(强的松)合用。若应用上述药物4~6周后效果不明显,可加用依托泊苷(足叶乙甙,Vpl6)100/(m2·d),静滴,1次/d,用3~5天,每月1个疗程。亦可采用巯嘌呤(6-MP)或硫鸟嘌呤(6-TG)每天60~75mg/m2口服。甲氨蝶呤(MTX)每次15~20mg/m2,每周1次口服或静脉注射。多脏器病变化疗时间一般不短于1年。若于用药后很快退热,精神食欲好转,1~2周后皮疹消退,肝、脾缩小,肺部症状减轻说明对治疗敏感,但骨骼X线改变常需数月方可恢复,肺部病变消退较慢。有些病人在治疗过程中可有反复或发生新的病状,则应改换其他药物,如环磷酰胺10mg/(kg·d),连续3天,每3周1次。
(3)近年来主张用足叶乙甙(VP16) 150mg/m2 静脉滴注,或 300mg/m2 口服,连用三天,每 3~4 周一个疗程共 6 个月。其毒副作用较小, 疗效也较好。
(4)其他常用化疗药物::长春花碱 (Vinblastin) 5~6.5mg/ (m2w) 静脉注射;甲氨喋呤(MTX)30mg/m2,每周 1~2 次; 6-巯基嘌呤(6-MP)2.5mg/(kgd);环磷酰胺 2.5~5mg/(kgd)等。
4、免疫治疗 胸腺素或胸腺肽,每次3~5mg静脉或肌内注射,连用2~3个月,可与化疗联合使用,增加疗效。近年来开始试用 α-干扰素(α-Interferon)和环胞菌素 A(cyclosporin A)对调节免疫功能, 减少化疗的远期副作用有较好的效果。
5、支持治疗 重型患者应住院并予最大剂量抗生素,保持气道通畅,营养支持(包括高能营养),血制品,皮肤护理,理疗以及必要的医护关怀.
6、对症治疗 包括用磺胺甲噁唑(SMZ)预防卡肺猪囊尾蚴病及积极控制感染,特别是中耳炎。如合并呼吸道感染导致呼吸衰竭,应及时给氧并需监护。对尿崩症患者应给加压素(垂体后叶激素)治疗。皮肤和牙龈损害.施行清创术,甚至可切除严重受损的牙龈组织,以限制口腔病变.头皮脂溢性样皮炎可使用含硒质洗发液(每周2次).若洗发液无效,可局部少量使用皮质类固醇药剂。继发侏儒的患儿可试用生长激素。此外预防出血、纠正贫血亦很重要。
预后
预后决定于发病年龄,受累器官的数目和有无脏器功能衰竭,受累器官少的,即使年龄较小亦有自然痊愈的可能,尤其是单纯皮肤浸润,对化疗反应好的预后亦佳,治疗缓解后亦有复发的病例,甚至数年后还可复发,年龄小于2岁,大于4个器官受累,伴有器官功能不全者,如不治疗病死率可高达90%以上。
小儿郎格罕细胞性组织细胞增生症中医治疗
当前疾病暂无相关疗法。
以上提供资料及其内容仅供参考,详细需要咨询医生。
日常保健
多补充一下体内缺乏的维生素营养物质。
病因和机理尚未明确,重点积极防治感染性疾病。
1、避免接触有害因素 避免接触有害化学物质尤其是药物,注意合理用药, 接触毒物或放射性物质时,应加强各种防护措施。
2、大力开展防治各种感染性疾病,尤其是病毒性感染。
3、加强体育锻炼,注意饮食卫生,保持心情舒畅,劳逸结合,增强机体抵抗力。
1.血象 多无特征性改变,一般以正细胞正色素性贫血为多见,重症者可有血小板减少或白细胞减少。
2.骨髓检查 LCH患者骨髓增生程度多正常,少数可有增生减低,骨髓内很少发现pLCs,因此骨髓细胞学检查缺乏特异性,一般在血象异常时方作为常规检查。
3.肝肾功能 肝肾功能检查异常提示预后不良,肝功能检查包括SAST,SALT,碱性磷酸酶和血胆红素增高,血浆蛋白减低,凝血酶原时间延长,纤维蛋白原含量降低等,肾功能检查包括尿素氮,肌酐,尿渗透压等,如有尿崩症症状还应检测尿相对密度和限水试验。
4.肺功能 如血气分析出现明显的低氧血症,则提示肺功能受损,肺功能不全常提示预后不良。
5.免疫学检查 常规免疫学检查多正常,T细胞亚群检查T4,T8均可减少,可有淋巴细胞转化功能降低,T淋巴细胞缺乏组胺受体。
6.病理活检或皮疹印片 病理活检见到典型的郎格罕细胞是诊断的依据,皮疹,淋巴结,骨病变处病灶局部穿刺物或刮出物均可行病理检查,其特点为有分化较好的组织细胞增生,也可见到泡沫样细胞,嗜酸粒细胞,淋巴细胞,浆细胞和多核巨细胞,不同类型可有不同的细胞组成,严重者可致原有组织破坏,但无分化较差的恶性组织细胞,慢性病变中可见大量含有多脂质性的组织细胞和嗜酸细胞,形成嗜酸细胞肉芽肿,增生中心可有出血和坏死。
7.免疫组织化学染色 S-100蛋白染色阳性,CD1a阳性,此外α-D-甘露糖酶,ATP酶和花生凝集酶也可呈阳性反应。
8.电子显微镜检查 病变组织内可见Langerhans巨细胞,该细胞为胞体不规则的巨大的单个核细胞,直径可达13µm,胞浆中可见分散的细胞器,以及称为Langerhans颗粒或Birbeck颗粒的特殊细胞器,支持本病的诊断,颗粒长190~360nm,宽33nm,末端可呈泡沫样扩张,形如网球拍,胞核不规则,呈扭曲状,核仁明显,多为1~3个。
9.骨骼X线检查 全身骨骼摄片可发现,病变特征为溶骨性破坏,扁平骨和长骨发生溶骨性骨质破坏,扁平骨病灶为虫蚀样至巨大缺损,形状不规则,边缘可呈锯齿状,颅骨巨大缺损可呈地图样,脊椎骨受压则呈扁平椎,但椎间隙不狭窄,长骨多为囊状缺损,单发或互相融合成分房状,骨皮质变薄,无死骨形成,破坏明显处可有层状骨膜增生,上下颌骨破坏可致牙齿脱落,以上典型的骨X线变化提示郎格罕细胞性组织细胞增生症。
10.肺部X线检查 双肺可有弥漫的网状或点网状阴影,可见局限或颗粒状阴影,需与粟粒样结核相鉴别,严重病例可见肺气肿或蜂窝状肺囊肿,纵隔气肿,气胸或皮下气肿。
湿疹样,皮脂溢出样皮疹, 可留有白斑或色素沉着;肺部广泛浸润,常并发呼吸道感染,极易发生肺炎,并发气胸和皮下气肿,呼吸衰竭是致死的主要原因;骨骼病变可出现病理性骨折,也可同时有溶骨性骨骼病变;发生椎弓破坏者,常伴神经压迫症状,如肢体麻木,疼痛,无力,瘫痪,甚至大小便失禁;可并发消化道溃疡;肝,脾,淋巴结增大;耳流脓,营养不良,生长发育迟滞,腹泻和贫血;并发尿崩症,胆道闭锁及上腔静脉综合征等。