尼曼-皮克病(Niemann-Pick disease,NPD)又称鞘磷脂沉积病,属先天性糖脂代谢性疾病。其特点是全身单核巨噬细胞和神经系统有大量的含有神经鞘磷脂的泡沫细胞,临床以肝、脾肿大和中枢神经系统受累为主要特点。本病为常染色体隐性遗传病,多见于两岁以内的婴幼儿,亦有新生儿时期发病的。
-
挂什么科:儿科 小儿内科
-
需做检查:尿常规 脾脏超声检查 骨髓象分析 血常规 血液生化六项检查 肝脏超声检查 腹部平片 视力 眼底检查
-
治疗方法:药物治疗 康复治疗
-
常用药物:暂无相关信息
-
一般费用:根据不同医院,收费标准不一致,市三甲医院约(6000——50000元)
-
传染性:无传染性
-
治愈周期:1-3个月
-
治愈率:无特效治疗方法
-
患病比例:发病率约为0.001%
-
好发人群:父母近亲结婚的婴幼儿
-
相关症状:
-
相关疾病:
1.A型: 在出生6个月内出现肝脾大,继之很快进展的中枢神经系统退化,早期神经系表现为肌张力低及肌无力,反映在喂养困难,淋巴结病及肺浸润,皮肤呈黄棕色,惊厥,半数患儿眼底黄斑区有樱桃红斑,常有生长发育迟缓,呼吸道合并症,多于2~3岁死亡,酸性鞘磷脂酶活性低于正常值的5%。
2.B型: 最常见,病情较A型轻,在儿童早期有肝脾大,腹部膨隆,重症患者肝受累可导致肝硬化,门脉高压及腹水,脾大可发展为脾功能亢进,全血细胞减少,应做部分或全脾切除术,轻症患者的脾大可到成人才被发现,在患者的X光胸片可见到弥漫性网状后结节状浸润,重症患者可导致慢性肺疾病,肺心病,多数患者无神经系受累,智力正常,也有个别报道有智力低下,寿命可活至成人,酸性鞘磷脂酶活性为正常值的5%~10%。
3.C型: 临床表现多样化,典型的表现为在儿童期有不同程度的肝脾大,垂直性核上性眼肌麻痹及缓慢进展的中枢神经系退化(进行性共济失调,张力障碍及痴呆),垂直性核上性眼肌麻痹为神经系脑干受累的标志,是有特征的体征,患儿在上幼儿园及小学时表现行为异常,常被认为笨,逐渐痴呆,构音障碍,语言困难,流口水,张力障碍首先表现在走路或跑步时手足的姿势异常,易摔跤,逐渐变为全身性,常伴有惊厥,青春期可发生精神障碍。
4.其他变异型: 致死性新生儿肝病,肝功能衰竭,婴儿早期进行性神经系统损害,伴有或无肝受累,成人伴有精神病或进行性痴呆。
(一)发病原因
本病征为先天性糖脂代谢异常性疾病,其特点是全身网状内皮系统有大量的含有神经鞘磷脂的泡沫细胞。
(二)发病机制
本病征是一种常染色体隐性遗传性疾病,约1/3病例有明显家族史,由于神经鞘磷脂缺乏致神经鞘磷脂代谢障碍,使脂与辅酶的作用失调,引起类脂质在体内过多沉积,在网状内皮系统中,出现肝,脾大,中枢神经系统退行性变,神经鞘磷脂是由N-酰基硝氨醇与1个分子的磷酸胆碱在C1部位连接而成,神经鞘磷脂酶来源于各种细胞膜和红细胞基质等,在细胞代谢衰老过程中被巨噬细胞吞噬后,经神经鞘磷脂酶将其水解成N-酰基鞘氨醇和磷酸胆碱,正常肝脏中此酶活力最高,脾,肾,脑和小肠降低至50%以下,本症分成 6型,儿童期以A,B,C三型为主。
A型及B型是由于ASM基因突变造成ASM活性减低,在单核巨噬细胞系统贮积了鞘磷脂及其他脂质,ASM基固定位于11p15.1~p15.4,已发现了12种突变型。
而C型则为细胞转运外源胆固醇的缺陷及溶酶体中贮积了未脂化的胆固醇,此型的分子缺陷还不清楚,在患者肝,脾组织中贮积物除未脂化的胆固醇外还有鞘磷脂,磷脂及糖脂,而脑组织中贮积物只有糖脂,白细胞中ASM活性正常,仅在经培养的细胞中,可有部分鞘磷脂酶缺乏,说明为继发于溶酶体中胆固醇的贮积,许多组织中可见到泡沫样细胞及海蓝组织细胞,这些细胞并不是C型的特异细胞,在一些无脏器肿大的病例中可没有,在皮肤及结膜活检可见到特异的包涵体。
诊断
诊断依据:
1.肝脾肿大。
2.有或无神经系统损害或眼底樱桃红斑。
3.外周血淋巴细胞和单核细胞胞浆有空泡。
4.骨髓可找到泡沫细胞。
5.X线肺部呈粟粒样或网状浸润。
6.有条件可作神经鞘磷脂酶活性测定,尿神经鞘磷脂排泄量及肝,脾或淋巴结活检证实。
鉴别诊断
1.戈谢病婴儿型 以肝大为主,肌张力亢进,痉挛,无眼底樱桃红斑,淋巴细胞胞浆无空泡,血清酸性磷酸酶升高,骨髓中找到戈谢细胞。
2.Wolman病 无眼底樱桃红斑,X线腹部平片可见双肾上腺肿大,外形不变,有弥漫性点状钙化阴影,淋巴细胞胞浆有空泡。
3.GM神经节苷酶脂病Ⅰ型 出生即有容貌特征,前额高,鼻梁低,皮肤粗,50%病例有眼底樱桃红斑和淋巴细胞胞浆有空泡,X线可见多发性骨发育不全,特别是椎骨。
4.Hurler病(黏多糖Ⅰ型) 肝脾大,智力差,淋巴细胞胞浆有空泡,骨髓有泡沫细胞等似NPD,心脏缺损,多发性骨发育不全,无肺浸润,尿黏多糖排出增多,中性粒细胞有特殊颗粒,6月后外形,骨骼变化明显,视力减退,角膜混浊。
小儿尼曼-皮克病西医治疗
目前医学手段尚无对此病有有效治疗方法。
1、一般治疗:以对症治疗为主,加强营养。控制饮食的摄入。
2、药物治疗:
A)使用抗氧化剂如维生素
C、E或丁羟基二苯乙烯,可阻止神经鞘磷脂M所含不饱和脂肪酸的过氧化和聚合作用,减少脂褐素和自由基形成。
B)C型患儿可试用二甲基亚砜。
D)基因重组酶替代治疗A、B型患儿正在研究中。
3、脾切除:适于非神经型、有脾功能亢进者。但需考虑脾切除后对患者的影响。
4、胚胎肝移植 已有成功的报道。
预后
本病征预后不良,多于2岁内死亡,A型或C型于发病后4年内死亡,B型与成人非神经型皆无神经系统症状,智能正常,可活至成年。C型及新斯科夏型大多死于神经系统的退行性变或继发呼吸道感染。
小儿尼曼-皮克病中医治疗
当前疾病暂无相关疗法。
以上提供资料及其内容仅供参考,详细需要咨询医生。
日常保健
1、多以清淡食物为主,注意饮食规律。
2、根据医生的建议合理饮食。
避免近亲婚配,做好遗传性疾病的咨询工作,测定皮肤成纤维细胞酶活性可以检出A和B型的半合子,培养羊水细胞酶活性检测可供做A,B型的产前诊断。
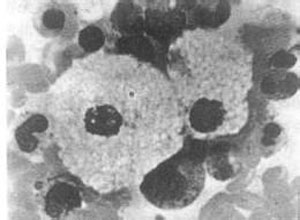
1.血象: 血红蛋白正常或具有轻度贫血;脾亢明显时白细胞减少,单核细胞和淋巴细胞常显示特征性空泡,约8~10个,具有诊断价值(图1),电镜下这些空泡系充满类脂的溶酶体,血小板数正常,晚期有脾亢和骨髓明显侵犯时可减少,患者白细胞缺乏神经磷脂酶活性。
2.骨髓象: 含有典型的尼曼-匹克细胞,常称泡沫细胞,该细胞直径20~100μm;核较小,圆形或卵圆形,一般为单个,也可有双核;胞浆丰富,充满圆滴状透明小泡,类似桑葚状或泡沫状,电镜下显示小泡周围有部分膜层结构环绕,用位相显微镜对未染色标本作检查,可显示细胞胞浆内呈小泡状,与戈谢细胞不同,在偏光下观察,小泡呈双折射性;在紫外线下荧光呈绿黄色,生化特点PAS反应弱阳性,胞浆内的小泡壁呈阳性,小泡中心阴性;酸性磷酸酶,碱性磷酸酶,过氧化物酶,苏丹黑均呈阴性反应。
3.血生化检查: 胆固醇,总脂可升高,SGPT轻度升高。
4.尿液检查: 排泄神经鞘磷脂量明显增加。
5.肝,脾和淋巴结活检: 均有成堆,成片或弥漫性神经鞘磷脂的泡沫细胞浸润。
6.鞘磷脂酶活性测定: 白细胞或培养的成纤维细胞鞘磷脂酶活性,各型酶的活性不同,对诊断最为可靠。
7.X线检查: 无特征性X线表现,在长期存活病例,由于充脂性组织细胞在骨骼内大量增殖可表现骨质疏松,髓腔增宽,骨皮质变薄,甚至长骨可出现局灶性破坏区,但无骨骼膨大畸形改变。
婴儿期以后肺泡受充脂性组织细胞浸润,肺部可见类似组织细胞增生X症的表现,肺部呈粟粒样或网状浸润,总之无特异性,仅提供辅助诊断的依据。
8.B超检查: 可见肝,脾,淋巴结肿大。
9.脑电图: 有异常脑波。
10.眼底检查: 可见樱桃红斑。
肝脾淋巴结肿大及慢性肺疾病,肺心病,惊厥,生长发育迟缓,可导致肝硬化,肝功能衰竭,发生门脉高压及腹水,发展为脾功能亢进,全血细胞减少,进行性共济失调,张力障碍及痴呆,伴有反复肺部感染,青春期可发生精神障碍。