粘多糖沉积是由于溶酶体酶缺陷造成酸性粘多糖不能降解,使组织中大量粘多糖沉积和尿中粘多糖排泄增加而导致的遗传性溶酶体病。根据不同的酶缺陷和不同的临床表现分为多种类型。不同的类型之间差别很大。临床表现涉及多个脏器,以智力障碍、体格发育障碍、眼部病变为最常见表现。
1、粘多糖贮积症I型(mucopolysaccharidosis)
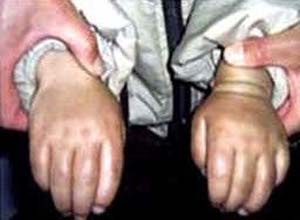
又称Hurler综合征,承雷病,AR,为溶酶体a-L-艾杜糖醛酸酶缺乏,发病率1/10万新生儿,主征;进行性发育迟缓,体矮,智力低下,舟状头,颈短,面容粗陋,有角膜混浊,关节僵硬,活动受限,驼背,其他:视网膜色素沉着,耳聋,胸廊畸形,肝脾肿大,腹大,心脏瓣膜缺损和动脉硬化等,粘多糖筛查阳性,根据酶活性测定可检出携带者,预后差,多于儿童期死亡。
2、粘多糖贮积症II型(mucopolysaccharidosis)
又称Hunter综合征,XR,无女性患者,发病率为6/10万,为艾杜糖醛酸硫酸酯酶缺乏,主征:与I型相似,但无角膜混浊,背不驼,其他:多毛,进行性耳聋;肌肉痉挛,行为莽撞,10岁后2/3患者有惊厥发作,生化诊断如I型,主要根据临床表现相鉴别,重型者多在青春期前死亡。
粘多糖沉积病是由于细胞溶酶体酸性水解酶先天性缺陷所致,其特征是过多的寡聚糖堆积与排泄。
诊断
名称 代号 酶缺陷 酶学测定样品 生化改变 遗传特性 Hurler,Scheie综合征 MPSⅠ α-L-艾杜糖酸苷酶 成纤维细胞,白细胞,组织,羊水细胞 尿和组织中DS,HS增多,成纤维细胞中DS增加 常染色体隐 性 Hunter综合征 MPSⅡ 艾杜糖醛酸硫酯酶 血清,成纤维细胞,白细胞,组织,羊水,羊水细胞 同上 X连隐性 Sanfilippo综合征A MPSⅢA HS-N-硫酸酯酶(硫酰胺酶) 成纤维细胞,白细胞,组织,羊水细胞 HS在尿中和组织中增多,DS在成纤维细胞中增多 常染色体隐性 Sanfilippo综合征B MPSⅢB α-N乙酰葡萄胺苷 血清,成纤维细胞,白细胞,组织,羊水细胞 HS出现于尿中 同上 Sanfilippo综合征C MPSⅢC 乙酰基转移酶 成纤维细胞 HS出现于尿中 同上 Morquio综合征 MPSⅣ N-乙酰半乳糖胺-6-硫酸酯酶 成纤维细胞 KS和CS出现于尿中 同上 Morquio综合征 β-半乳糖甘酶 成纤维细胞 KS出现于尿中 Maroteaux-Lamy综合征 MPSⅥ N-乙酰半乳糖胺4-硫酸酯酶(芳香硫酸酯酶B) 成纤维细胞,白细胞,组织,羊水细胞 DS出现于尿中 同上 β-葡糖醛酸苷酶缺乏症无名疾病 MPSⅦ β-葡糖醛酸苷酶 血清,成纤维细胞,白细胞,羊水细胞 DS,HS(±)出现于尿中 同上 无名疾病 MPSⅧ N-乙酰葡糖胺6-硫酸酯酶 成纤维细胞 KS和HS(±)出现于尿中 同上
注:MPS-粘多糖沉积症DS-磷酸皮肤素
HS-硫酸乙酰肝素CD-硫酸软骨素
KS-硫酸角质素
遗传性粘多糖沉积症约占出生婴儿的1/30000。
鉴别诊断
要鉴别呆小症,佝偻病等病。
呆小症表现为生长发育过程明显受到阻滞,特别是骨骼系统和神经系统,如:(1)身体矮小,上身长,下身短,并常伴有四肢骨畸形等,因为甲状腺激素和主长激素一样是长骨生长和骨骼正常发育所必需的因素,(2)表情淡漠,精神呆滞,动作迟缓,智力低下,并常可有耳聋,这主要是由于神经细胞树突与轴突的形成,髓鞘与胶质细胞的生长,神经系统机能的发生,发展,脑的血流供应等,均有赖于适量的甲状腺激素,甲状素激素缺乏则导致这一系列过程的障碍,(3)常伴有体温偏低,毛发稀少,面部浮肿等一系列甲状腺功能低下的一般症状,呆小症患儿出生时身高,体重等可无明显异常,至3~6个月时,则出现明显症状,如果能在出生3个月左右即明确诊断,开始补充甲状腺素,可以使患儿基本正常发育,一旦发现过晚,贻误了早期治疗时机,则治疗难以生效。
佝偻病诊断:
(1)一般症状:早期出现睡眠不安,夜惊,好哭,烦躁,病情进展后可见全身肌肉松弛,肝脾大,腹部突出,多汗,贫血,发育迟缓等。
(2)头部:颅骨软化,多见于6个月以内的小儿,在顶骨或枕骨中心用手指按压,有乒乓球样弹性感觉,方颅,前囟门特大,闭合延迟(可延至2岁以上),出牙晚,10个月以上还不生乳牙,且牙质不坚。
(3)胸部:肋骨与肋软骨交界处膨大,称为“肋骨串珠”,胸骨前突,胸腔前后径增大,称为“鸡胸”,胸廓沿膈肌附着处内陷成沟,称为郝氏沟。
(4)脊柱:多向后凸,偶为侧弯。
(5)四肢:腕部尺,桡骨骺端膨出,成钝圆形隆起,称佝偻病性手镯,下肢弯曲,形成X形腿,O形腿,军刀腿等。
粘多糖沉积病西医治疗
这个病迄今为止没有很好的办法,骨髓移植可以做,但疗效并不是如想像中的那么好,目前世界上最先进的治疗是一种Enzyme Replacement Therapy。酶替代和基因治疗法正在研究中。据文献统计,国外已有几十例靠骨髓移植治疗而成功的例子。
粘多糖沉积病中医治疗
当前疾病暂无相关疗法。
饮食清淡富于营养,注意膳食平衡。忌辛辣刺激食物。以免造成病情反复的情况,比如说,海鲜、鸡肉、狗肉等。与此同时,也不要禁食刺激性的食物。多吃新鲜的蔬菜和水果。新鲜的蔬菜和水果含有大量人体所需的营养成分。多吃提高免疫力的食物,以提高机体抗病能力。
粘多糖沉积病是由于细胞溶酶体酸性水解酶先天性缺陷所致,除了遵从婚姻法规定,避免近亲结婚,减少后代遗传病发病率外,无有效预防措施。
据文献统计,国外已有几十例靠骨髓移植治疗而成功的例子,如果手术成功的话可以成功解除脏器衰竭的危险,虽然孩子的智力发育,骨骼生长可能会比正常同龄人稍逊,但是过正常人的生活还是没有问题的。
粘多糖沉积病是由于细胞溶酶体酸性水解酶先天性缺陷所致,根据病因目前本病可分为八大类型,我国已报告200从余例,主要表现为严重的骨骼畸型,肿脾肿大,智力障碍以及其它畸形,粘多糖沉积病产前诊断以测定培养羊水细胞内特异的酶活力最为可靠,但实验要求高,一般实验室难开展,两种较简单的实用的方法是甲苯胺蓝定性及糖醛酸法半定量测定。
1.甲苯胺蓝定性:方法同尿液粘多糖检查,妊娠早期羊水正常者可呈阳性,妊娠中后期为阴性如阳性提示胎儿患粘多糖沉积病。
2.糖醛酸半定量测定:羊水中酸性粘多糖与四硼酸钠硫酸溶液反应生成糖醛酸,以每毫无肌酐中糖醛酸的量反映酸性糖多糖的多少,随着妊娠的进展,糖醛酸含量逐渐减少,妊娠16-20周的参考值为3.3~7.0mg/mgCr,如高于此值应考虑有粘多糖沉积肥病,本法除Morguio综合征外对其它类型的粘多糖沉积病均有诊断意义。
3.常染色体隐性遗传病(autosomal recessive disorder):致病基因在常染色体上,基因性状是隐性的,即只有纯合子时才显示病状,此种遗传病父母双方均为致病基因携带者,故多见于近亲婚配者的子女,子代有1/4的概率患病,子女患病概率均等,许多遗传代谢异常的疾病,属常染色体隐性遗传病,按照“一个基因,一个酶”(one gene one enzyme)或“一个顺反子,一个多肽”(one cistron one polypeptide)的概念,这些遗传代谢病的酶或蛋白分子的异常,来自各自编码基因的异常,常见的常染色体隐性遗传病有溶酶体贮积症,如糖原贮积症,脂质贮积症,粘多糖贮积症;合成酶的缺陷如血γ球蛋白缺乏症,白化病;苯丙酮尿症,肝豆状核变性(Wilson病)及半乳糖血症等。
视网膜色素沉着,耳聋,胸廊畸形,肝脾肿大,腹大,心脏瓣膜缺损和动脉硬化等。