黏多糖贮积症Ⅶ型又称戈尔伯杰(Goldberg)综合征,β-葡萄糖苷酸酶缺乏症。其特征为智力正常或略落后、骨骼改变,特殊面容、侏儒、肝脾肿大和有疝气,尿中可以排出过多硫酸皮肤素,为一种常染色体隐性遗传病,虽然生化缺陷相同,但各例的临床表现和起病年龄皆有不同。
生后不久就出现特殊容貌,肝脾肿大和生长迟缓,以后可出现反复呼吸道感染,反应迟钝,驼背和其他畸形,面容粗陋,两眼间距增宽,扁平鼻梁,上颌突出,角膜周边可有细微的基质改变,也有些病人角膜正常,骨骼异常变异性很大,可进行性加重,也可为轻度到中度改变,有多发性骨发育不全,脊柱后弯,鸡胸,肋骨下缘外翻,矮小,肝脾肿大,有脐疝或腹股沟疝,可多次发生呼吸道感染,有明显的智力低下,部分病人可有主动脉狭窄,所有病人均无耳聋,部分病人可有脑积水,初生即确诊的患儿尚有新生儿黄疸和血小板减少,以后可有严重发育落后和智力低下。
(一)发病原因
本病的病因是常染色体隐性遗传。
(二)发病机制
本病的发病机制是β-葡萄糖苷酸酶缺乏,基因定位在7q21.23~q22,除硫酸角质素外,所有酸性黏多糖都含有葡萄糖苷酸末端部分,并且这一末端部分以β-构形,与乙酰或硫酸氨基己糖末端结合,在这些酸性黏多糖水解时,因受内葡萄糖苷酸酶(玻璃酸酶)或外葡萄糖苷酸酶连续酶促,结果可使β-葡萄糖末端露出,为了使外葡萄糖苷酸酶进一步分解β-葡萄糖苷酸链,就必须有β-葡萄糖苷酸酶作用,黏多糖贮积症Ⅶ病人体内缺乏β-葡萄糖苷酸酶,故发生部分水解的硫酸软骨素,硫酸皮肤素等就会沉积于组织细胞内而致病。
根据临床症状,X线表现和实验室检查,就可考虑为黏多糖贮积症Ⅶ型,再进一步通过细胞培养发现β-葡萄糖苷酸酶缺乏,即可确诊。
黏多糖贮积症Ⅶ型西医治疗
无特殊疗法。严重畸形者可酌情给予手术矫形,反复呼吸道感染和心力衰竭者,均应施行有效的对症治疗。
预后
较好,大多可存活到成人。
黏多糖贮积症Ⅶ型中医治疗
当前疾病暂无相关疗法。
(以上提供资料及其内容仅供参考,详细需要咨询医生。)
一旦发现糖原累积症,以防治低血糖为主,膳食少量多餐,限制脂肪和总热量,限制体力活动。
血清乳酸高者,宜服碳酸氢钠防治酸中毒。皮质激素、肾上腺素、胰高血糖素等可帮助控制低血糖。
糖原累积病是一组儿童遗传性糖原代谢紊乱的疾病,主要以机体组织糖原累积过多而分解困难为特点,极少为糖原合成代谢障碍,致使机体组织糖原储存少,糖原累积病并非为一种疾病,而是一组疾病,目前确定的已有12种,临床均以低血糖为特征,所涉及到的器官主要为肝,肾,骨骼肌,遗传方式多为常染色体隐性遗传,无性别差异,多在儿童期发病,部分病人至成人后,病情不再发展,可以维持一般健康水平。
患者主要是由于缺乏分解糖原的某些酶,如葡萄糖-6-磷酸酶,α-1,4葡萄糖甙酶,磷酸果糖激酶,肝磷酸化激酶等。
许多患者的父母为近亲结婚,故要避免近亲结婚预防本病,一旦发现糖原累积症,以防治低血糖为主,膳食少量多餐,限制脂肪和总热量,限制体力活动,血清乳酸高者,宜服碳酸氢钠防治酸中毒,皮质激素,肾上腺素,胰高血糖素等可帮助控制低血糖。
尿中过多排出的黏多糖为硫酸皮肤索,硫酸软骨素也可有不同程度的增加,中性粒细胞和淋巴细胞有异染性颗粒。
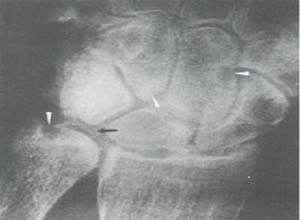
X线检查:X线表现为多发性成骨不全,上肢骨变短,伴有肱骨近端髓腔扩大,骨骺发育显示成熟加速,但骨膜新骨形成却很少,有胸骨,管状骨增粗等,椎体变扁平而小,脊柱后凸,椎间隙相对增宽,椎体形态完整,没有舌状突出,前肋变宽,肋骨下缘外翻,髂骨小,髂骨嵴有不规则透亮区,边缘硬化,肩胛骨下缘亦有类似改变,长骨干骺端和髋臼不规则,可能与佝偻病相混淆。
本病可并发肝脾肿大,脐疝或腹股沟疝,多次发生呼吸道感染,明显的智力低下,主动脉狭窄,脑积水,尚有新生儿黄疸和血小板减少。