常染色体隐性小脑性共济失调(autosomal recessive cerebellar ataxias) 这类疾病中,较重要的有Friedreich共济失调、共济失调毛细血管扩张症等,本处重点讨论前者。
Friedreich共济失调(Friedreichs ataxia,FA)是常染色体隐性遗传病,临床特点是儿童起病,进行性共济失调、心肌病,下肢深感觉丧失、腱反射消失,以及锥体束征,常伴骨骼畸形。现已知本病累及多个系统,临床表现复杂多样,随着致病基因被克隆以及发现基因有GAA三核苷酸重复扩展,对本病的发病机制有了新的了解。
-
挂什么科:儿科 小儿内科
-
需做检查:肌电图 脑电图检查 脑脊液检查-化学检查-蛋白质检查 神经系统核医学检查 颅脑MRI检查 神经系统检查 小气脑CT扫描
-
治疗方法:手术治疗 药物治疗 支持性治疗
-
常用药物:暂无相关信息
-
一般费用:根据不同医院,收费标准不一致,市三甲医院约(10000 —— 20000元)
-
传染性:无传染性
-
治愈周期:3个月
-
治愈率:40%
-
患病比例:0.002%
-
好发人群:儿童
-
相关症状:
-
相关疾病:
1.典型病例
起病年龄2~16岁,平均11岁,绝大多数在20岁以前起病,首发症状为躯干和下肢共济失调,步态不稳,跑步困难,Romberg征阳性(两下肢并拢不能站稳),以后累及上肢,表现为震颤,指鼻试验阳性,轮替运动不良等,少数病例以脊柱侧弯,肢体笨拙或心脏病为首发症状,早期不一定有构音障碍,锥体束征或深感觉减低或消失,数年后这些症状都相继出现,跟腱和膝腱反射消失,多数病人上肢腱反射也消失或减弱,双侧巴氏征阳性但肌张力不高,下肢振动觉和位置觉减弱或消失,触觉减退,痛,温觉正常,2/3以上病人有脊柱侧弯,严重者影响心肺功能,常见弓形足或内翻足见图1,晚期可见肢体远端肌肉萎缩和无力,下肢较上肢明显,晚期还可见视神经萎缩,白内障,眼球震颤,少数病人有感觉神经性耳聋,眩晕,病晚期智力发育迟缓,心理过程减慢和情绪不稳等均不少见。
心肌病常为进行性,心律不齐,心力衰竭可在共济失调症状以后出现,也可在以前出现,心电图异常在神经症状出现以前即可测知,可见T波倒置,ST段下降,QRS波幅低或心律失常,心脏扩大,有杂音,超声心动图显示肥厚性心肌病,晚期有心力衰竭,此外,糖尿病或糖耐量曲线异常约见于10%~20%的病人,一般在30~40岁时明显。
体感诱发电位不论病期或病情轻重,均为异常,肌电图可见肌束震颤,MRI可见脊髓萎缩,上颈段明显,PET在尚能行走的病人可见脑局部葡萄糖代谢率比正常为高,而在晚期已不能行走的病人则局部代谢率降低。
2.不典型的Friedreich共济失调
常可见到,可能是由于不同的等位基因,也可能是其他疾病,确诊常需靠基因分析。
(1)迟发型Friedreich:30岁左右起病,进展较慢,症状较轻。
(2)腱反射保留的FA:15岁以前起病,膝,踝腱反射存在,早期心肌病,病死率高。
(3)伴有维生素E缺乏的FA:有典型FA的临床症状,维生素E缺乏。
(4)不伴心肌病,骨骼异常,肌萎缩的病例。
(5)MRI显示脊髓变性轻而小脑变性重的病例。
(6)共济失调伴眼球运动失用症:常染色体隐性遗传,进行性小脑性共济失调,腱反射消失,周围神经病,眼球运动失用症,脊柱侧弯,内翻足,1~15岁起病,寿命较长。
(一)发病原因
本病是常染色体隐性遗传,少数为散发,致病基因(FRDA)定位于9q13,FRDA的突变形式,少数为点突变,而98%表现为GAA三核苷酸重复扩展,正常人GAA扩展的数目各民族不同,在7~22或5~10之间,FA病人的GAA扩展数目可达200~900,与正常人的重复数目无重叠,GAA重复次数与起病年龄呈负相关,起病3~20岁者,重复数为800~900;30岁左右起病者,重复数为201~734,伴发糖尿病或肥厚性心肌病者,重复次数较多,近来也报道有例外的情况:有些典型临床症状而无GAA扩展;而有些GAA扩展但无FA的典型临床症状(McCabe等,2000)。
(二)发病机制
基因突变导致基因产物,即线粒体蛋白frataxin的减少,脊髓和心肌是frataxin表达最高的组织;肝,骨骼肌和胰腺的表达中等,在基因突变时,基因表达水平最高的组织最先受累,因而脊髓变性和心肌病是FA最主要的表现,至于组织病变的基本原因还不完全清楚,有认为,frataxin直接影响线粒体的能量代谢和氧化磷酸化作用,也有认为frataxin对线粒体铁的转运有调节功能,认为本病的发生与细胞内铁的分布异常有关,线粒体内有铁沉积,从而诱导氧自由基产生而导致细胞损伤,也有人认为本病的发生与肌醇磷脂代谢异常有关,影响神经冲动的突触传导。
本病的病变广泛,最突出的病理见于脊髓,脊髓后柱,脊髓小脑束,锥体束,后根均可见髓鞘脱失和轴突变性,脊髓腰骶部受累最重,小脑皮质有变性,齿状核,下橄榄核,前庭核,脑桥核也有不同程度变性,有报道大脑皮质运动区有轻微神经元改变,心肌病是本病的特征之一,常是进行性的心肌肥厚,慢性间质性纤维变性和炎性浸润。
诊断
根据临床症状和家族史,可以做出初步诊断,但由于本病的表型差别很大,最准确的诊断要靠DNA检测,在DNA分析之前,可参考Harding(1981)的临床诊断标准以做出可能的诊断:
1.儿童起病。
2.隐性遗传。
3.进行性躯干和下肢共济失调。
4.下肢腱反射消失。
5.逐渐出现构音障碍,锥体束征,深感觉障碍,肢体无力。
6.心肌病。
7.10%伴发糖尿病,或糖耐量异常。
8.约2/3有脊柱侧弯,弓形足。
9.少数出现远端肌萎缩,视神经萎缩,白内障,眼震。
鉴别诊断
本病需与小儿时期起病的其他遗传性慢性进行性共济失调作鉴别。
1.共济失调毛细血管扩张症 有毛细血管扩张,免疫缺陷,无骨畸形,无感觉障碍。
2.无β-脂蛋白血症 有棘红细胞增多,脂肪泻,血脂减低。
3.Refsum病 有夜盲,视网膜色素变性,鱼鳞癣,血清植烷酸增高。
4.遗传性痉挛性截瘫 膝腱反射亢进,可伴视神经萎缩,智力低下。
5.Marinesco-Sjorgren综合征 有先天性白内障,智力低下。
小儿常染色体隐性小脑性共济失调西医治疗
本病无特殊治疗。可对症处理。手术治疗脊柱侧弯应慎重,如果侧弯超过40°仍能行走的病儿,可考虑手术,术前应监测心肺功能。心肌病严重者治疗心力衰减。有糖尿病者可试用胰岛素,但多无效。病之早期应尽量做平衡训练和锻炼肌力,做理疗。现有作者提出试用抗氧化剂、铁螯合剂、自由基清除剂等药物,尚无经验总结。
预后
病情进展缓慢,在起病6~27年以后不能独立行走。起病越早,不能行走也越早。心肌病和糖尿病是预后不良的指征,是大多数病人的死亡原因。
小儿常染色体隐性小脑性共济失调中医治疗
当前疾病暂无相关疗法。
以上提供资料及其内容仅供参考,详细需要咨询医生。
重视产前诊断,必要时终止妊娠。
治疗相当困难,开展产前诊断是减少发病的关键,可采用长PCR技术检测GAA反复数目即可做出基因诊断,携带者诊断和产前诊断,必要时应终止妊娠。
1.脑脊液检查 病儿脑脊液检查正常,偶有蛋白轻度增高和细胞数轻度增多。
2.血糖检查 血糖,糖耐量检验不正常。
3.肌肉活检 可见直径大的神经纤维脱髓鞘及轴索断裂,以及非特异性的神经性肌萎缩。
4.DNA检测 由于98%的FRDA基因可测得GAA重复扩展,故可采用长PCR技术检测GAA反复数目即可做出基因诊断,携带者诊断和产前诊断,如果只在一个等位基因上有GAA重复扩展(杂合子),则需进一步检查另一等位基因有无点突变。
5.心电图检查 可见Q-T间期延长或T波倒置,心律不齐。
6.肌电图检查 显示感觉神经传导速度减慢,感觉神经传导速度在下肢消失,在上肢减慢,肌电图可见失神经性异常。
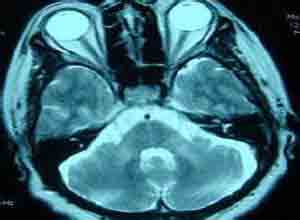
7.CT,MRI检查 头颅CT检查正常或轻度异常,MRI可见脊髓变细,萎缩,小脑和脑干萎缩。
8.诱发电位检查 诱发电位有异常,视觉诱发电位可有潜伏期延长,波幅降低,提示有轴突变性,但临床可无视觉症状,听觉诱发电位自乳突电极记录可能降低或消失,认为与螺旋神经节变性有关,临床上不一定有听觉症状,体感诱发电位均为异常。
可发生脊柱侧弯,弓形足或内翻足,肢体远端肌肉萎缩和无力,肢体笨拙,构音障碍,锥体束征或深感觉减低或消失,视神经萎缩,白内障,智力发育迟缓,心理过程减慢和情绪不稳等,可发生心律不齐,心力衰竭,发生糖尿病等。