地中海贫血(Beta thalassaemia)(简称地贫)又称海洋性贫血(thalassemia),据全国医学名词审定委员会规定应称为“珠蛋白生成障碍贫血”。是由于一种或多种珠蛋白肽链合成受阻或完全抑制,导致Hb成分组成异常,引起慢性溶血性贫血。根据不同类型的珠蛋白基因缺失或缺陷,而引起相应的珠蛋链合成受抑制情况不同,可将地贫分为α-地中海贫血;β-地中海贫血,δ-地中海贫血、γ-地中海贫血及少见的β-地中海贫血;以前2种类型常见,各类地中海贫血之间又可互相组合,可与各种异常Hb组合(如HbE/β地中海贫血),这一组疾病又称地中海贫血综合征,均属常染色体不完全显性遗传。
-
挂什么科:内科 血液科
-
需做检查:骨髓象分析 心电图 嗜碱性点彩红细胞 有核红细胞 便常规 涂片
-
治疗方法:药物治疗 支持治疗
-
常用药物:暂无相关信息
-
一般费用:根据不同医院,收费标准不一致,市三甲医院约(3000——10000元)
-
传染性:无传染性
-
治愈周期:1-3个月
-
治愈率:35%
-
患病比例:0.0001%
-
好发人群:婴幼儿
-
相关症状:
-
相关疾病:
患儿出生时无症状,多于婴儿期发病,生后3~6个月内发病者占50%,偶有新生儿期发病者,发病年龄愈早,病情愈重,严重的慢性进行性贫血,需依靠输血维持生命,3~4周输血1次,随年龄增长日益明显,伴骨骼改变,首先发生于掌骨,再至长骨,肋骨,最后为颅骨,形成特殊面容(Downs面容):头大,额部突起,两颧略高,鼻梁低陷,眼距增宽,眼睑水肿,皮肤斑状色素沉着,食欲不振,生长发育停滞,肝脾日渐肿大,以脾大明显,可达盆腔,患儿常并发支气管炎或肺炎,并发含铁血黄素沉着症时因过多的铁沉着于心肌和其他脏器如肝,胰腺等而引起该脏器损害的相应症状,其中最严重的是心力衰竭和肝纤维化及肝功能衰竭,是导致患儿死亡的重要原因之一,本病如不治疗,多于5岁前死亡。
(一)发病原因
1.病因
β珠蛋白基因位于11号染色体短臂1区2带(简记:11P12),本病除少数几种为几个核苷酸缺失外,绝大部分都是点突变(单个核苷酸置换,增加或缺失)所致,全世界已发现100种基因突变类型,我国有20种(表1),突变致β链合成部分受抑制者称“β 地中海贫血”;致β链完全受抑制者称“βo地中海贫血”,肽链合成的抑制涉及δ链,称δβ海洋性贫血(δβ 或δβo),染色体上的2个等位基因突变点相同者称纯合子;等位基因的突变不同者称“双重杂合子”;同源的染色体上只有1个突变者称“杂合子”。
2.分类
根据β基因缺陷所产生的杂合子和纯合子的不同,其临床表现亦有差异,按照病情轻重的不同,可将β地中海贫血为重型,轻型和中间型三种类型。
(1)重型β地中海贫血:重症β地中海贫血β-mediterranean anemia major)又称库理贫血(Cooleys anemia),这是β地中海贫血纯合子,β和β 地中海贫血双重杂合子或β 地中海贫血纯合子或δβo纯合子,是我国常见的一种地中海贫血,本症患者双亲均为β-地贫杂合子,其子女中获得重症β-地贫几率为25%,50%为杂合子,余25%为正常。
(2)中间型β地中海贫血:中间型β海洋性贫血(β-mediterranean anemia intermedia)包括:β 基因纯合子,某些β0/β 双重杂合子,非典型β地贫杂合子,合并α地贫血和δβ地贫,其临床表现介于重型与轻型之间,特点:
①发病年龄较晚(常于4~5岁)。
②中度贫血,Hb 60~90g/L。
③轻度肝脾肿大。
④外周血涂片红细胞形态与重型类似。
⑤HbF含量增高,HbA2可稍高,正常或降低。
⑥输血量小或不必输血仍可维持生命;家系调查证实为上述基因传递。
非典型β地贫杂合子:指位于β珠蛋白基因远侧的启动区域位点控制区(locus control region,LCR),这些并非在β链上的分子缺陷(非典型β地贫基因)与典型的β地贫基因组合形成双重杂合子。
(3)轻型β-地中海贫血:轻型β-地中海贫血(β-mediterranean anemia minor或trait)是β0,β 或δβ基因,包括双重突变杂合体(指病变的β珠蛋白基因的核苷酸序列上同时有2种突变点)的杂合子状态,如[TATA box-28(A→G)CDl7(A→T)/N],主要表现为轻度小细胞,低色素贫血,尤其婴儿期明显,可有轻度黄疸和脾大。
(二)发病机制
β-地贫的许多病理生理和临床表现均与珠蛋白链合成不平衡有关,由于β链的合成受到抑制,故HbA(α2β2)的合成可减少或不存在,在杂合子,多余的α链与代偿性增多的δ链结合,使AbA2(α2δ2)增加,在纯合子由于β链的显著减少,α链相对增加,多余的α链与γ链结合,故HbF(α2γ2)成为红细胞中主要的Hb成分,由于HbF较HbA的氧亲合力高,在组织中不易释出氧,故患者常有组织缺氧,缺氧引起红细胞生成素的大量分泌,刺激骨髓造血功能,红骨髓极度扩张,因而引起一系列骨骼改变。
由于α与β链之间的不平衡,过剩的α链可聚合成极不稳定的α2,α3,或α4,易变性沉积于幼红细胞和红细胞中而形成α链包涵体,由于包涵体附着于红细胞膜而使红细胞膜变僵硬,易受机械性损伤,在骨髓内破坏而不能全部进入血液循环,导致“无效造血”,部分含有包涵体的红细胞虽然成熟并释放至外周血,但这些红细胞通过微循环时,易被破坏,使其寿命缩短,此外,红细胞的包涵体还影响红细胞的通透性,进一步使其寿命缩短,导致溶血性贫血。
贫血促进肠道对铁的吸收增加(达80%),加以铁利用障碍和治疗过程中的反复输血,使心,肝,脾,骨髓及皮肤等组织沉积大量的铁,导致含铁血黄素沉着症,晚期产生继发性血色病,心肌和肝功能等遭受损害,糖尿病和其他内分泌障碍等。
根据临床表现和血液检查特别是HbF含量增高及家系调查可确诊,有条件者可进一步作肽链分析或基因诊断。
鉴别诊断
1.缺铁性贫血:需注意与发生于婴儿期重症缺铁性贫血区别。
2.血红蛋白E病:HbE/β海洋性贫血综合征与本病相似,但前者Hb电泳可见HbE>30%。
3.红细胞G-6PD缺乏所致CNSHA 重型者与重型β海洋性贫血临床表现相似,但前者感染及氧化性药物可加重贫血,红细胞Heinz小体阳性,HbF含量正常可鉴别。
小儿β地中海贫血西医治疗
(一)治疗
1.治疗原则
轻型地中海贫血不需治疗;中间型α地中海贫血应避免感染和用过氧化性药物,中度贫血伴脾肿大者可做切脾手术,中间型β地中海贫血一般不输血,但遇感染,应激,手术等情况下,可适当予浓缩红细胞输注;重型β地中海贫血,高量输血联合除铁治疗是基本的治疗措施;造血干细胞移植(包括骨髓,外周血,脐血)是根治本病的惟一临床方法,有条件者应争取尽早行根治手术。
2.输浓缩红细胞
(1)低量输血:单纯的输血或输红细胞最终导致血色病,中等量输血疗法,使血红蛋白维持在60~70g/L,实践证明,这种输血方法虽然使重型患者有望摆脱近期死亡的威胁,但患者的生存质量随年龄增长越来越差,相当一部分患者于第2个十年内因脏器功能衰竭而死亡。
(2)高量输血: ①高量输浓缩红细胞的优点:纠正机体缺氧;减少肠道吸收铁;抑制脾肿大;纠正患儿生长发育缓慢状态, ②方法:先反复输浓缩红细胞,使患儿血红蛋白含量达120~140g/L,然后每隔3~4周Hb≤80~90g/L时输注浓缩红细胞10~15ml/kg,使Hb含量维持在100g/L以上。
3.铁螯合剂
因长期高量输血,骨髓红细胞造血旺盛,“无效红细胞生成”以及胃肠道铁吸收的增加,常导致体内铁超负荷易合并血色病,损害心肝,肾及内分泌器官功能,当患者体内的铁累积到20g以上时,则可出现明显的中毒表现,故应予铁螯合剂治疗, 1岁内使用铁螯合剂,其副作用如骨骼畸形,生长抑制的发生率明显升高,一般主张2~3岁后或患儿接受10~20次输血后并有铁负荷过重的证据,血清铁(SF)>1000μg/L,血清转铁蛋白完全饱和才开始除铁治疗,当前临床广泛使用的是去铁胺(Deferoxamine,DFO),剂量:20~50mg/(kg·d),加注射用水或生理盐水用便携式输液泵每天(或每晚)腹壁皮下注射8~12h,每周连用5~6天,用药前后应作SF,尿铁的监测,若SF>3000μg/L或者有铁负荷继发心脏病时,可予DFO 50~70mg/(kg·d)持续24h静脉滴注,使用铁整合剂时加用维生素C口服可增加尿中铁的排泄量1倍,但维生素 C可将铁从储备部位动员出来并通过氧化代谢间接影响心肌细胞,故在重度铁负荷时不宜使用大剂量维生素 C,一般每天口服100~200mg,在停用DFO期间也不应坚持服维生素 C,长期使用DFO一般无明显的毒副作用,注射局部反应,皮疹,疼痛,无需停药,但铁负荷轻者使用大剂量DFO可出现白内障,听力丧失,长骨生长障碍等,应引起临床重视,Johon等对47例地贫患者接受DFO治疗的毒副作用研究发现,DFO大剂量与SF<2000μg/L是引起DFO毒性的两大危险因素,提出治疗指数(TI),即平均每天DFO剂量(mg/kg)除以血清铁蛋白浓度(μg/L),可指导临床给药,当TI<0.025时,一般无毒性,近十年来,国外一系列新型口服铁螯合剂如defefipone(L1),多价阴离子胺(the poly anionicanine,HBED),多价氮替代物(the substituted poly aza compounox TR coll),PIH等相继问世,在动物实验中已证实长期服用能有效地降低机体铁负荷,但只有L1试用于人体,通过高量输血与除铁治疗可维持患者正常生长发育及达到正常人的生活质量及寿命,但必须终身承受沉重的经济负担,可能的输血相关合并症及心理负担。
4.造血干细胞移植(HSCT) HSCT
是当前临床上根治本病的惟一方法,HSCT包括骨髓移植(BMT),脐血移植(UCBT),外周血造血干细胞移植(PBSCT)和宫内造血干细胞移植(IUSCT),迄今,全世界已成功开展HSCT 1200例,其中BMT达1000余例,PBSCT 10例,UCBT约30例,IUSCT 2例,研究发现,重型地贫的HSCT有其自身的特点。
(1)受体的选择:移植前病者3个危险因素评分标准将其分为3类:Ⅰ类:零分,Ⅱ类:1~2分,Ⅲ类:3分,危险因素评分: ①去铁胺应用史: A.“0”分为规则使用,即经1次规则输血后18个月开始,每周至少5天,皮下输注持续8~10h, B.“1”分为不规则使用,未达上述任一标准, ②肝大: A.“0”分为肝活检无纤维化, B.“1”分为纤维化, BMT效果顺序为Ⅰ>Ⅱ>Ⅲ类;无病存活率分别为85%,80%,53%,纤维化及铁负荷是重要危险因素。
(2)供体选择: ①血缘相关HLA全相合的供体:同胞HLA相合供体BMT可治愈80%的重型β地贫,但仅25%~30%患者可找到HLA相合的家系成员供体,作者等自1998年1月国内首例亲属UCBT治疗地贫取得成功以来,至今共完成HSCT 12例,植入率83.3%,5年无病存活率(EFS)58.3%, ②血缘相关HLA不全相合(包括同胞,双亲)或单倍体相合供体:Polchi等报道18例患者BMT后生存率及无病生存率分别为58%和26%,排斥率41%,总病死率44%,Gaziev等报道29例结果:植入率44.8%;排斥率55%,相关病死率34%,因此,找不到HLA相合供体,且无法接受输血及去铁治疗者,才考虑进行此类移植, ③非血缘相关(UD)供体:Dihi报道3例地贫UD-BMT:1例植活,1例回复地贫状态,1例死于移植物抗宿主病(GVHD);Miano报告8例,4例成功,3例排斥,1例死亡,发生排斥是此类移植面临的主要问题,2000年6月,作者等以非血缘相关UCBT治疗1例重型地贫患儿,取得了成功,现患儿已脱地贫状态存活180天, IUSCT近年已有成功报道,1996年Touraine等用胎肝造血干细胞宫内移植治疗2例宫内确诊为地贫胎儿,1例死亡,1例生下无病生存已达4年之久,但Monni等用父亲骨髓CD34细胞经胎儿腹腔注射治疗1例胎龄为10周的重型B地贫胎儿未获成功,目前IUSCT成功所需单个有核细胞数,移植的最佳胎龄,植入后的状态等尚需进一步深入研究。
5.脾切除,大部分脾栓塞术
(1)重型β地中海贫血: ①脾切除指征:重型β地贫伴脾功能亢进者,行脾切除能减少对输血的需要量,并减轻体内铁负荷,但切脾仅是姑息疗法,而且,切脾面临着发生严重感染等致命并发症的危险,故脾切除应有严格的指征: A.输血量 B.脾功能亢进:红细胞破坏增加,持续的白细胞或血小板减少, C.巨脾引起压迫症状, D.一般年龄应在5岁以上, Maniga等提出输血商(TQ)患者每年每公斤体重的输血量除以同龄维持同一Hb水平,脾脏正常患者每年每公斤体重的输血商>1,可作为临床判断早期脾功能亢进的指标, ②脾大部分栓塞治疗(PSE):由于脾脏为重要的免疫器官,为避免脾切除后继发性免疫功能低下和凶险的感染,有学者提出脾大部分栓塞治疗(PSE)代替脾切除,Politis等对比了PSE与脾切除术后5年的追踪随访资料,结果显示两组病人输血量较前都有减少,但PSE组血中IgM浓度明显高于脾切除组,且感染的发生率也明显低于后者,但有资料显示PSE对中间型β地贫效果较满意,对重型β地贫的疗效不满意,可能与重型β地贫粗大颗粒的红细胞包涵体主要在骨髓破坏有关,以上2种手术后均可导致肝脏代偿性溶血,引起明显肝大,纤维化加重。
(2)中间型仅地中海贫血(HbH)病:中度/重度贫血(Hb<80g/L)无黄疸的HbH患病者,行切脾术疗效极佳,可使Hb上升至90~110g/L,若术前Hb>80g/L或慢性溶血性黄疸者,切脾常无效。
6.基因治疗
从分子水平上纠正致病基因的表达,即基因治疗,其途径有2:
(1)将正常的β珠蛋白的基因导入患者的造血干细胞,以纠正β地贫的遗传缺陷,必须解决以下3个难题: ①转移的外源珠蛋白基因能在细胞和整体达到高度表达, ②必须分离,纯化获得用于基因传导的人类造血干细胞, ③α基因与β基因之间表达协同一致性,此外,转导的外源性基因必须随珠蛋白基因系统在个体发育过程中适时表达仍处于实验研究阶段。
(2)采用某些药物调节珠蛋白基因的表达:采用某些药物调节珠蛋白基因的表达以平衡α,β珠蛋白的肽链水平,目前临床应用以调节珠蛋白基因表达的药物有白消安(马利兰),羟基脲,丁酸盐,阿糖胞苷(Ara-C),红细胞生成素(Epo)和异烟肼(雷米封)等,其中羟基脲(Hu)应用及实验研究较多,羟基脲(Hu)是一种低毒和可有效增加γ珠蛋白链和β珠蛋白链合成,从而导致血液学和临床症状明显改善,羟基脲(Hu)治疗的剂量及方法: ①5天疗法:50mg/(kg·d)×5天为1个疗程, ②10~30mg/(kg·d),连用3周为1个疗程,或25~50mg/(kg·d)×5~7天为1个疗程, ③有学者采用15~20mg/(kg·d)连续用药方法,主要对某些β地贫基因缺陷类型有效: A.-28/654-2或-28/41-42双重杂合子,β-28纯合子, B.IVS-2-654 C→T突变中间型β-地贫, C.HbE/β-28双重杂合子, 5~7天显效,Hb上升水平20~45g/L,中间型效果明显,重症者一般用药初期效果明显,随治疗时间延长,效果渐差,人们正对这种药物调控基因的机制深入研究,从而加深对珠蛋白基因表达的遗传调控的认识。
(二)预后
β-地中海贫血重型并发症常是导致患儿死亡的重要原因,如不治疗,多于5岁前死亡,中间型和轻型地中海贫血正确处理可长期存活。
小儿β地中海贫血中医治疗
当前疾病暂无相关疗法。
以上提供资料及其内容仅供参考,详细需要咨询医生。
1.高蛋白低脂肪:对一般贫血病人来说,首先应考虑给予高蛋白饮食。这可以通过食用动物的瘦肉以及肝,肾等内脏,获得优质蛋白的补充。其次,应尽量控制脂肪的摄入量,因为脂肪可抑制人体的造血功能,高脂肪还可导致腹泻、消化不良、肥胖病等疾患。
2.丰富的维生素:饮食结构中维生素的含量丰富,对各类疾病的患者都是适宜的。就贫血病人而言,维生素B1,维生素B12,维生素C和叶酸等是至关重要的。维生素B1的补充,可以通过粮食特别是粗杂粮食物获得;维生素 B12和叶酸主要来源于动物内脏等食物;维生素C的主要来源则是各种新鲜的蔬菜和水果。
积极开展优生优育工作,以减少/控制“地中海贫血”基因的遗传。
1.婚前地中海贫血筛查,避免轻型地中海贫血患者联婚,可明显降低重型/中间型地贫患者出生的机会。
2.推广产前诊断技术,对父母双方或一方地贫基因携带者,孕4个月时,采集胎儿绒毛,羊水细胞或脐血,获得基因组DNA以聚合酶链反应(PCR)技术对高危胎儿进行产前诊断,重型/中间型患儿应终止妊娠。
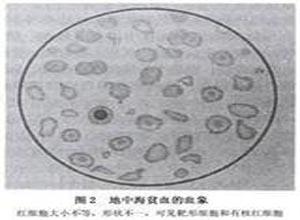
1.血象
Hb 100~120g/L,红细胞<2.0×1012/L,红细胞大小不等,呈小细胞低色素性贫血,中央浅染,外周血涂片红细胞异形明显,可见梨形,泪滴状,小球形,三角形,靶形及碎片,嗜碱性点彩红细胞,多嗜性红细胞,有核红细胞增加,网织红细胞增加(≤0.1),白细胞及血小板数增加,并发脾功能亢进。
2.骨髓象
有核红细胞增生极度活跃,粒∶红比值倒置,以中,晚幼红细胞为主,胞体小,核固缩,胞浆少偏蓝,甲基紫染色可见晚幼红细胞含包涵体(仅链沉淀)。
3.红细胞盐水渗透性试验
红细胞渗透脆性减低,0.3%~0.2%或更低才完全溶血。
4.HbF测定
这是诊断重型β珠蛋白生成障碍性贫血的重要依据,HbF含量轻度升高(<5%)或明显增高(20%~99.6%);HbA2常降低,正常或中度增高,HbA2 3.5%~8.0%。
5.肽链分析
采用高效液相层析分析法可测定α,β,γ,δ肽链的含量,Cooley贫血时,β/α比值<0.1(正常值为1.0~1.1),因本病多为点突变,故常用PCR加ASO才能明确突变点,我国各民族的β地贫基因突变情况有一定差异,南方汉族的突变基因以CD41-42(-TCTTT),CDL7(A→T),IVS-Ⅱ-654(C→T)和TATA-box28(A→G)为主,占85%~90%,双重杂合子的突变组合可达近100种。
6.其他
血浆及红细胞内维生素E含量显著下降,与病情呈正相关;超氧阴离子自由基增加。
常规做X线,B超,心电图等检查。
骨骼X线检查,骨髓腔增宽,皮质变薄和骨质疏松,颅骨的内外板变薄,板障加宽和短发样骨刺形成。
可并发黄疸,肝脾肿大,胆石,可并发溶血危象,水肿,腹水,贫血,骨骼改变,生长发育停滞,常并发支气管或肺炎,并发含铁血黄素沉着症,造成脏器损害,并发心力衰竭,肝纤维化,肝功衰竭等。