铁粒幼红细胞性贫血(sideroblastic anaemias,SA)是由一组病因不同的血红素合成障碍和铁利用不良所引起的贫血。特点是骨髓中出现大量“环形”铁粒幼红细胞,且铁剂治疗无效。本病分为获得性和遗传性,获得性又分为原发性和继发性。
遗传性或获得性铁粒幼红细胞性贫血的临床表现均相似。病程发展多缓慢呈良性过程,常以贫血表现来就诊。有皮肤黏膜不同程度的苍白或轻度黄疸,半数有脾或肝大但较轻,故有时不能触及。出血表现仅偶见。
临床症状:
1.贫血为本病主要症状与体征。常有皮肤苍白,部分病人皮肤呈暗黑色。软弱,动则心悸、气促。肝脾轻度肿大。
2.发生血色病时(即含铁血黄素沉积症)肝脾肿大显著。发生血色病时可出现心、肾、肝、肺功能不全,少数可发生糖尿病。
3.由于长期贫血导致严重营养不良、智力发育落后、免疫力低下。
临床分型:
1.遗传性:男性发病,女性为携带者。少数常染色体遗传者男女均可发病。早期症状为软弱,乏力。贫血多为中度,但差别很大,甚至有的亲属中贫血程度也不相同。
2.获得性:男女均可发病。主要表现为贫血,由药物引起的贫血可相当严重,甚至需要输血。发病缓慢。
(1)原发性:起病隐匿,病情进展缓慢。以贫血为主要表现,贫血程度轻重不一。
(2)继发性:患者除有本症的特点外,尚有原发病的症状或有应用药物的病史。
(一)发病原因
遗传性:分为性联遗传或常染色体隐性遗传。
1.X染色体伴性遗传
是最多见的类型。这种家族中只有男性出现贫血,女性携带者一般无贫血,但红细胞群常呈明显的双相性。还有的家族中子代只有女性患病,可能为X连锁显性遗传,因男性不能生存,而无男患者出现。目前在X伴性遗传方面尚未完全定论。
2.常染色体遗传
有些家系表现为父子间的垂直遗传,另有些家族发现兄弟姐妹之间同患本病,且程度也相同,提示遗传方式为常染色体隐性遗传或常染色体显性遗传。获得性:获得性又可按病因分为原发性和继发性。 原发性:原因不明,多见于青壮年。虽然原因未明,但能完全排除接触有害的化学或物理因素的可能性。 继发性:各种细胞毒药物,例如治疗恶性肿瘤与白血病药物如氮芥、环磷酰胺。6-巯基嘌呤、阿糖胞苷、氨甲喋呤和阿霉素等。其他化学因素如苯、油漆、汽油、农药等也与再障发生有关。各种电离辐射,急、慢性感染,包括细菌(伤寒等)、病毒(肝炎、EBV、CMV、B19等病毒)、寄生虫(疟原虫等)。也可继发于各种血液疾病。 某些疾病可导致铁代谢异常,如骨髓异常增生综合症、白血病、淋巴瘤、和其它系统的癌症、类风湿性关节炎、卟啉病、恶性贫血、吸收不良综合症、溶血性贫血、粘液性水肿等,这些疾病可引起铁代 谢异常,而并发铁粒幼细胞贫血。 不论遗传或获得性患儿均以对吡哆醇(维生素B6)治疗反应的效果而进一步定为效应性或无效性,其中以遗传性者对维生素B6反应较佳。
(二)发病机制
遗传性
在血红素生物合成过程中,某些酶缺乏或生化过程中产生障碍时,即可导致幼红细胞内非血红素铁的过量蓄积,产生病态性铁粒幼红细胞。在红细胞的线粒体中,作为卟啉生物合成的第一步,甘氨酸与琥珀酸辅酶A结合成δ-氨基γ酮戊酸(ALA)。吡哆醇(维生素B6)则在体内转变成具有生物活性的5-磷酸吡哆醛(PLP),后者作为ALA合成过程不可缺少的辅酶参与这一反应。有证据表明本病的发生是由于这一生化过程存在着遗传缺陷,而各家族的生化缺陷不一定相同。
获得性
PLP的拮抗物能抑制骨髓细胞的RNA合成,更重要的是抑制线粒体的蛋白质,包括某些细胞色素和细胞色素氧化酶的合成。可能与线粒体中的细胞色素的损害相关,可抑制吡哆醇转变为PLP,可以抑制ALA脱水酶血红素合成酶及抑制粪卟啉原Ⅲ转变为原卟啉Ⅲ,因而在慢性铅中毒时可发生铁粒幼细胞贫血。
病理改变
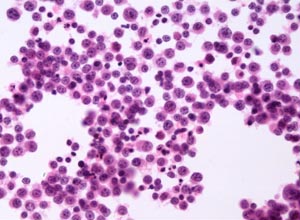
正常人骨髓的幼红细胞胞浆内可见少量铁存在,用铁染色时可见30%~60%的幼红细胞中有细小的铁粒,但数量仅1~2粒,最多不超过5粒。而本病的幼红细胞则不但含粗大的铁粒,数量多且质呈现病态。在光镜下可见铁粒在幼红细胞核的周围呈环状或至少1/3环形。在电镜下此种异常的线粒体变形、肿胀、破裂,铁以尘状或斑状沉积于线粒体的内嵴,失去铁蛋白或含铁血黄素的超微结构。由于铁沉积,也抑制了中幼红细胞进入细胞增殖周期,以致出现红细胞无效造血现象。
诊断:
根据根据发病年龄,本病的特点,一般不难作出铁粒幼细胞贫血的诊断。
诊断依据:
1.低色素性贫血,网织红细胞不高。
2.骨髓红细胞系增生,骨髓涂片用铁染色后出现大量“环形”铁粒幼红细胞。
3.血清铁含量增高,铁饱和度大于正常。诊断一般并不困难,误诊和漏诊多由于未作骨髓铁染色检查所致。患儿若近期有服药史或正在服药中,则应详问其药物名称、用量及其原发病,如服用易并发本病的药物或制剂,于停用后,症状大多可较快消失,试行停药亦可作为本病继发于药物或毒物的反证诊断。
鉴别诊断:
1.两种铁粒幼细胞贫血相鉴别,必须根据详细的病史与体检综合考虑,以及进行家族调查。获得性常有明显的原发病因可寻。
2.与其他贫血类型鉴别 缺铁性贫血:缺铁性贫血是体内铁的储存不能满足正常红细胞生成的需要而发生的贫血。是由于铁摄入量不足、吸收量减少、需要量增加、铁利用障碍或丢失过多所至。形态学表现为小细胞低色素性贫血。 地中海贫血:有家族史,血片可见大量靶形红细胞,血红蛋白A2增加,血清铁蛋白及骨髓可染铁增加。 慢性炎症性贫血:总铁结合力正常或降低,血清铁蛋白增高。
3.与其他表现类似的疾病鉴别 遗传性SA容易MDS中RAS相混淆,应注意鉴别。MDS是一组干细胞异常综合征,造血系统表现为红系、粒系和巨核细胞系分化紊乱的形态学特点,即所谓“病态造血”表现。贫血为大细胞性或正细胞正色素性,而遗传性SA多为低色素小细胞性贫血,无病态造血表现,除红细胞系外,其他系造血细胞无异常。此外,RAS仅少数对维生素B6治疗有反应,而遗传性SA应用大剂量维生素B6治疗部分病例有效。
小儿铁粒幼红细胞性贫血西医治疗
遗传性及获得性的患儿,都应首先试用大量维生素B6治疗,不论早期效果如何亦均应较长期服用。约半数病人有不同程度的疗效,一般每天剂量为50~200mg。复发后再用维生素B6(吡哆醇)治疗有时仍有效,但往往疗效不如第一次,有的则变为无效。
对维生素B6治疗无效的病例,可试用磷酸吡哆醛或色氨酸。亦可试用叶酸、维生素B12、肝精(肝浸膏)、丙酸睾酮、免疫抑制药或肾上腺皮质激素等。如已出现粒细胞发育不良的情况亦可试用小剂量阿糖胞苷(Ara-C)。
使用雄性激素,需要较长的治疗时间,故必须坚持应用2~4个月以上才能做出评价,有时要在治疗6个月后才出现疗效,病情缓解后仍应继续用药3~6个月再减量,维持1~2年。对儿童的疗效优于成人。适用于慢性轻、中度贫血的病儿。
对重度贫血者和用维生素B6(吡哆醇)无效者可输浓集的红细胞,处于婴儿期最好使血红蛋白值高于80g/L,以满足小儿生长发育基本需要。当血红蛋白下降至70g/L(7g/dl)左右,临床出现食欲不振、无力和有心衰可能时,才考虑输血。
体内如有过量的铁贮积,若病情不能耐受则可给予铁螯合剂治疗。可用去铁胺(去铁敏)等,输注去铁胺(去铁敏)50mg/(m2·d),同时加维生素C 100mg,每天连续输注8~16h,可减轻或推迟发生含铁血黄素沉着症。个别病人可以换部分血。
继发性病例则主要积极治疗原发病或停用有关的药物。先天性者可考虑异基因骨髓移植。
预后
本病的预后依其病因、病情轻重及对吡哆醇(维生素B6)治疗的反应而异。不论何种病因,对维生素B6的反应很重要。有7%~10%的获得性特发性病人可有发展为骨髓增生异常综合征(MDS)的趋向,最终转化为急性非淋巴细胞白血病,但过程急缓不一。大多数获得性特发性病人呈慢性无痛苦性过程。但如出现下列情况,常提示预后不良:如血小板减少,严重贫血;难以治疗的巨幼红细胞增多;获得性血红蛋白H的出现;骨髓中原始粒细胞增加;红细胞系统增生不良;复合性染色体核型异常或出现亚二倍体,若周围血中绝对性单核细胞增多或假性Pelger-Hut核异常均为向白血病转化的一种征兆,预后很坏。而血内如出现血小板数目增多,骨髓中明显红细胞系统增生时,可能预示预后较好。遗传性长期重症患儿可有血色病,皮肤呈棕铜色及糖尿病,肝、心功能异常等并发症。因药物所致,一般停药贫血即可纠正。如因恶性肿瘤血液所致预后较差。
临床上对铁粒幼细胞性贫血病症治疗效果的评价,需要从以下几个方面入手:
1、缓解:临床症状明显改善,血红蛋白明显升高,铁代谢实验室检查基本正常。环状铁粒幼红细胞明显减少。
2、进步:临床症状有所改善,血红蛋白有所提高,铁代谢实验室检查有所好转,环状铁粒幼红细胞减少。
3、无效:临床症状及实验室检查无进步。
小儿铁粒幼红细胞性贫血中医治疗
中医认为本病属于“内伤血虚”或“虚劳亡血”。按中医理论,肾主骨,骨生髓,故治疗宜从补肾着手,进行辨症施治。
南党参、熟地黄、炙黄芪、山萸肉、乌梅、茯苓、当归、陈皮、菟丝子、五味子、白术、山药、姜黄、丹参、吴茱萸、龙眼肉、炙甘草、阿胶等。出血加板蓝根、白茅根、生地、白芍、连翘、白及、三七;咽干减党参、黄芪,加金银花、白花、蛇舌草;腰膝酸软加旱莲草、杜仲、女贞子、山萸肉;腹胀呕吐加青皮、枳壳、半夏、佛手;纳呆苔腻加鸡内金、莱菔子、砂仁、藿香、佩兰叶。以上方剂每日1剂水煎服,30剂为一疗程。
以上提供资料及其内容仅供参考,详细需要咨询医生。
日常保健
改变不良的饮食习惯,尤其要做到不偏食不挑食和不长期素食。从食物中摄取足够的叶酸和维生素B12。多吃富含叶酸的绿色新鲜蔬菜和酵母类食物。多吃富含维生素B12的动物性食物。补充维生素C促进叶酸吸收。主要有绿色新鲜蔬菜、水果、花生仁、酵母、豆制品以及动物肝肾等。含维生素B12丰富的食物主要也是动物肝肾瘦肉。禁酒,有报告患慢性酒精中毒的病例多数伴有叶酸缺乏,故不应饮酒。
日常预防
1、注意合理用药,避免接触苯等有害化学物质。
2、孕期要注意各种不良因素的影响,做好相应的产检,有家族史的要进行遗传咨询。
1.血象:低色素性贫血,一般为中度贫血(血红蛋白在70-90g/L)少数为重度贫血(血红蛋白30-60g/L)。血红蛋白多在70~100g/L(7~10g /dl),偶亦可见低至30g/L(3g/dl)。遗传性所致者成熟红细胞多呈典型的、数量不等的低色素小细胞。获得性者则可同时见到正色素正常红细胞或大红细胞。网织红细胞反而减低,或增高不明显。白细胞、血小板计数一般正常,但在获得性病人中可有抑制情况。
2.骨髓检查:骨髓片中可见红细胞系统明显增生,以中幼红细胞为主,亦可有巨幼样变(对叶酸有反应者可见巨幼红细胞改变)。胞浆可见空泡、浆量少,缺乏血红蛋白形成,用普鲁士蓝染色出现病理性铁粒幼红细胞及“环形”铁粒幼红细胞,可达40%。粒系及巨核系正常。
3.血生化:血清铁含量增高,可达35.8μmol/L(200μg/dl);转铁蛋白饱和度增加,可达90%以上;血清总铁结合力和(或)不饱和铁结合力降低。血浆铁转换率增高。血中未结合的胆红素轻度升高。中性粒细胞碱性磷酸梅积分减低。
4.红细胞游离原卟啉:在遗传性者多减低,说明为ALA合成酶或粪卟啉原氧化酶缺陷。不论遗传性还是特发获得性病例中,当血红素合成酶或铁螯合酶有缺陷时,红细胞游离原卟啉均可增加。常规做B超和X线检查。
5.51Cr测出的红细胞生存时间正常或稍缩短,红细胞平均寿命40~100天。
6.尿中黄尿酸(48-二羟基喹林甲酸)和(或)犬尿喹啉酸的排泄增加,表示色氨酸的代谢异常。
7.红细胞内FEP减少或在正常下限,红细胞内FEC大多正常。吡哆醇治疗无效的病例,FEC可很高,而FEP显著减少。
铁过多是本病常见的并发症,可因铁大量沉积而合并血色病、糖尿病、肝硬化、心肌病等。在疾病晚期,可导致死亡。此外,铁过多可导致免疫功能低下,可因并发感染而致死。
常可见肝脾轻度至中度肿大,肝功能正常或轻度异常。重症可并发心力衰竭、肝功不全等。
约有1/3的患者出现糖尿病,偶见皮肤色素沉着。贫血严重的幼儿及少儿生长发育迟缓。
可并发消化道、泌尿道等出血。