淋巴瘤样肉芽肿(lymphomatoid granulomatosis,LG)是血管炎和肉芽肿反应的一种少见类型。本病的特征是各种组织均发生血管营养性,血管破坏性病变和肉芽肿反应,并伴以广泛的非典型性淋巴增殖性浸润,主要累及肺脏,也可累及肺外组织。
-
挂什么科:内科 呼吸内科
-
需做检查:皮疹 狼疮细胞 类风湿因子 尿免疫球蛋白(Ig) 尿常规 胸部平片 心电图 浆细胞 血常规 异型淋巴细胞 血液检查 中耳
-
治疗方法:药物治疗 支持性治疗
-
常用药物:暂无相关信息
-
一般费用:根据不同医院,收费标准不一致,市三甲医院约(5000——10000元)
-
传染性:无传染性
-
治愈周期:3个月
-
治愈率:40%
-
患病比例:0.002%
-
好发人群:无特殊人群
-
相关症状:
-
相关疾病:
LYG的临床表现与其他类型肺淋巴瘤有较大重叠,主要受累器官为肺(80%),皮肤(40%~45%),中枢神经系统(30%),肝(25%);其次为肾,消化道,骨髓和眼等,全身症状包括发热,周身不适,乏力,体重减轻,关节疼痛和皮肤损害等,通常在出现症状14个月内死于泛发性肺部病变和继发感染,因其主要侵犯肺部,故最常见的主诉为下呼吸道症状,如咳嗽,咯血,胸痛及呼吸困难,有30%出现神经系统损害,表现为精神错乱,共济失调,癫痫发作及颅神经功能障碍等,尸解发现肝,脾,淋巴结,肾,心脏和胃肠道均可受侵犯,肺外的主要表现在皮肤,可见于40%~45%病例,皮损主要为浸润性红色斑块及皮下或真皮结节,亦可发生溃疡,斑丘疹或红斑性损害,多对称发生于下肢和臀部,亦可泛发全身,皮损可先于肺部损害或同时发生,而且常伴全身症状,如发烧,倦怠,体重下降,肌痛及关节痛等,胸外症状常见,因不同部位受累而出现相应临床表现。
1.呼吸系统
呼吸道症状常为主诉,病人大多有咳嗽,咳痰,呼吸困难或气喘,胸痛等,肺部受累是本病最常见的早期表现,常伴有不同程度的发热,消瘦,少数患者也可只有肺部X线异常,而无临床症状,有些患者起病时就有肺部受累,呈进行性发展,另有一些患者也可在病程之中发生进行性肺部受累,病变主要位于两下肺野,尤以两肺外带为多见,肺尖部很少受累,病变几乎总是呈双侧性,并且波及范围很广,但也可为非对称性,虽然肺部病变具有淋巴瘤的特征,但很少有双侧肺门淋巴结肿大,淋巴结肿大仅发生在其他器官系统,而且表现也不典型,临床病情与淋巴瘤相似的患者,约1/3可出现肺空洞,可因大咯血而死亡,肺实质大片破坏引起呼吸衰竭也是本病的主要死亡原因,本病发生呼吸道受累的范围一般不大,但有时也可以呼吸道病变为主要临床特征,表现为阻塞性细支气管炎,这是由于大量炎性细胞浸润和纤维组织增生导致细支气管溃疡形成,破坏和闭塞所造成的,也可发生肺叶支气管部分阻塞,这是由于坏死组织和炎性细胞广泛侵犯支气管内膜所造成的。
肺实质内梗死样坏死,细胞高度浸润性结节,大块肺损害,广泛性肺实变及非典型性间质性肺浸润等,均可发生,非典型性淋巴组织细胞浸润侵犯肺动脉,肺静脉,偶尔也可引起这些脉管闭塞和广泛性肺实质坏死,此时可造成严重咳嗽和呼吸困难。
2.神经系统
表现有周围神经病,脑神经麻痹和各种中枢神经系统症状及体征,神经系统受累时可出现失语,头痛,感觉异常,偏瘫,共济失调,精神错乱,抽搐等,可发生于肺病变以前,中间,甚至肺部病变缓解时,周围神经病变多为非对称性,中枢神经系统病变可累及脑和脊髓的任何部位,可出现Bell麻痹,暂时性失明,复视,突眼,视力下降或眩晕,常见症状包括失语,轻偏瘫,失明,运动失调,截瘫,动眼神经麻痹,脑神经麻痹,头痛,感觉异常,意识模糊,昏迷,抽风,四肢瘫,视神经水肿,耳聋,面瘫,感觉迟钝,脑水肿,脑膜脑炎等,周围神经受累出现下肢感觉异常,可发生在其他系统病变出现之前,甚至当肺内病变在不断消退缓解时,中枢神经系统病变仍可在不断发展,一个患者多发生上述症状中的一种或数种,神经系统病变的发生与预后直接相关,一旦神经系统受累,其死亡率高达80%以上。
3.皮肤
肺外最常见受累部位是皮肤,为大片浸润性红斑,结节,溃疡,皮肤损害发生率占40%~50%,有10%~25%病人以首发皮肤损害出现;皮损可以先于肺脏受累2~9年出现,Liebow报告的一组40例患者,16例(42.5%)出现了皮肤损害,由于皮肤损害发生率高,活检容易以及病变的典型组织学所见,因此在疑诊淋巴瘤样肉芽肿时应仔细进行皮肤科检查,最常见的典型的皮损所见是1~4cm大小红紫色斑疹,丘疹或为硬而隆起的皮下结节(有时发生溃疡) ,直径2~3cm,多位于肢体,病损可发生于任何部位,但常见于臀部,腹部,大腿股部和下肢,修复过程常伴有瘢痕和色素沉着,其他皮损为非特异性改变,如小泡,广泛的鳞癣,斑状脱发,局部无汗和环状斑块,皮下结节有时很大,偶尔也可为其主要临床表现,主要皮肤损害也可表现为开始呈红色,最后转变成铜钱色,硬结状的皮下损害,皮肤损害通常与肺部病变同时发生,但可发生在肺部病变之前或之后数月到数年,有无皮肤损害与预后无关。
4.肾脏受累
约半数患者有肾脏组织学改变,但无临床症状,临床上明显肾脏受累罕见,而尸检可发现LG改变。
5.其他
其他病变10%的患者可有肝大,肝功能衰竭,少数病人有淋巴结肿大,脾大和腹水等,肝大也可为本病的早期表现,由于本病广泛浸润肝脏,可造成进行性肝功能衰竭而死亡,多数病例可出现淋巴结肿大,个别病例可出现巨脾,并伴有淋巴瘤样肉芽肿浸润,可引起白细胞减少。
除临床表现外,主要靠肺活检和皮肤活检证实有特征性病理改变,皮肤损害的病理改变与肺内改变相似,表现为非典型性淋巴细胞和浆细胞浸润,此改变主要位于真皮附属器周围,有些皮肤活检标本无血管炎或很难见到典型血管改变,但可见到其他改变。
(一)发病原因
病因不明,主要侵犯肺部,表现为坏死性肉芽肿和坏死性血管炎伴异形淋巴样细胞浸润,部分最后发展为淋巴瘤,有人认为是淋巴瘤的变形,从组织病理上看,本病既有血管炎改变,又有肉芽肿反应,这些改变符合变态反应,从而推测淋巴瘤样肉芽肿可能是一种伴有变态反应的肿瘤性疾病,伴有的变态反应可能是针对尚未证实的一种肿瘤抗原的抗原抗体反应,体内免疫反应失调或损伤性疾病,如肾移植,病毒性肝炎,干燥综合征及类风湿关节炎,常并发淋巴瘤样肉芽肿,这种相关作用及淋巴瘤样肉芽肿,与非典型性淋巴网状细胞浸润有一定的相似性,用慢性免疫抑制治疗某些先天性免疫缺损状态如Wishkott-Aldrich综合征,Louis-Bar综合征,偶尔也可并发淋巴瘤样肉芽肿,以上临床观察说明淋巴瘤样肉芽肿可能是一种继免疫失调或受损而发生的免疫增生性疾病,也有人考虑过自身免疫机制对淋巴瘤样肉芽肿的致病作用,另据部分患者存有T淋巴细胞功能不全,有肾移植后发生LYG的病例,以及LG和类风湿性关节炎或干燥综合征伴发,故考虑免疫异常和LYG的发生可能有关。
部分患者在病程中检出EB病毒感染的证据,嗜异性凝集试验阳性,故提示病毒可能参与其发病过程,此外,部分病例可发展为非霍奇金淋巴瘤中的血管中心性淋巴瘤(angiocentric lymphoma,AL),因此有作者认为LG是淋巴瘤前期阶段,甚至将全部LYG归入恶性淋巴瘤的少见特殊类型。
(二)发病机制
发病机制还不很清楚,有人认为是淋巴瘤的变形,表现为坏死性肉芽肿和坏死性血管炎伴异形淋巴样细胞浸润,就实验模型来说,慢性同种异体抗原刺激能激活宿主的慢性免疫活性细胞,并诱致非典型性淋巴增殖及淋巴样组织发生肿瘤,病毒对本病的发病可能起重要作用,它可通过改变宿主的膜抗原性而启动自身免疫反应,或通过致肿瘤病毒直接刺激网状内皮系统增生而造成非典型性或明显的淋巴网状细胞增殖,到目前为止,仍不能排除某些环境因素对本病的诱发作用。
抗原抗体反应,免疫失调,病毒感染和环境因素等,可能为单个因素,也可能为多种因素的综合作用,而导致本病的发生。
淋巴瘤样肉芽肿主要侵犯结外器官,累及的器官依次为肺(80%),皮肤(40%~50%),中枢神经系统(30%),肝(25%),肾,骨髓,眼等,而浅表淋巴结受累少见。
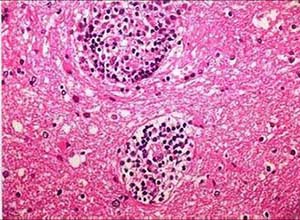
本病的病理特征表现为浸润细胞是由具有浆细胞样特征的非典型性淋巴网状细胞及少量中性粒细胞或嗜酸粒细胞组成的,非典型性淋巴网状细胞主要以成熟的淋巴细胞和浆细胞占优势,此类细胞比成熟淋巴细胞有较大的胞体和较多的细胞质,具有免疫母细胞的许多特征,常显示为有丝分裂相,本病血管炎性改变较明显,但并不一定同时具有几个系统的坏死性血管炎改变,有的有典型的白细胞破碎性或原发性纤维蛋白样坏死性血管炎,它是一种血管破坏性和血管营养性病变,主要累及中,小动脉和静脉,脉管病变是由非典型性淋巴网状细胞浸润所造成的,浸润细胞可使血管内皮隆起,血管内膜增厚,造成管腔狭窄,最后发生血栓性闭塞和广泛性血管坏死和血管重新形成,根据浸润细胞的类型及坏死范围可分为Ⅲ级,G1:主要由小淋巴,组织及浆细胞浸润,无不典型淋巴细胞及免疫母细胞,一般无坏死,G2:形态学介于G1和G3之间的过渡类型,G3:浸润细胞以非典型淋巴细胞为主,多形性的炎症背景常不明显,坏死较突出,G3又称血管中心性淋巴瘤,上述增生的淋巴细胞电镜及免疫学检测证实属外周T细胞,主要为T4,通常不能检出TCR基因重组。
本病出现的细胞浸润在组织学上表现为显著活跃增生,这就使病变具有了恶性变的特征,但它又与淋巴瘤不同,细胞浸润变化呈多样性,成熟与未成熟的淋巴样细胞常混合存在,可伴有广泛坏死性严重血管炎,甚至在距离浸润灶很远的部位也可见有坏死性血管炎,而浸润灶附近的淋巴组织却不受侵犯,这些都与淋巴瘤显著不同,然而,某些病例也可发展为免疫母细胞肉瘤或非典型性淋巴肉瘤,从而提示在本病与淋巴瘤之间可能存在一定的关系。
目前一致认为LG是一种独特的多形性细胞浸润形成的血管中心性淋巴瘤,PCR和原位杂交强烈提示EB病毒参与肺血管中心性淋巴瘤的发病,开始曾认为这些病变是周围T细胞淋巴瘤,因为免疫分型研究证实它是以表达T细胞相关抗原的不典型细胞为主,最近PCR和免疫组化证实有大的,不典型,EBV阳性B细胞单克隆亚群,提示很多LYG型血管中心性淋巴瘤实际上为EBV阳性B细胞淋巴瘤伴大量反应性T细胞,然而有些病变表现为周围T细胞淋巴瘤,这些肿瘤为EBV阴性,上述病理改变可累及气管,支气管,肺,脾,肾,脑及淋巴结。
肺脏常受侵犯,通常为多发性结节性浸润,病变大小不同,在梗死样坏死区偶有空洞形成,是本病肺部浸润的特征,有时病变也可为泛发性,同时伴有多发性肺脓肿,表现为一叶肺或一侧肺化脓性改变或破坏,Saldmm认为,本病发生的肺坏死与韦格内肉芽肿不同,前者主要是凝固性坏死,后者则为液化性坏死,某些病例尚可见到肺部机化性愈合区又发生完全性透明变性,还有些病例除有特征性血管向心性浸润外,尚可见有显著的成纤维细胞增生,但无非典型性淋巴网状细胞,并且坏死的程度也很轻,除广泛性血管炎和坏死外,常可见有由非典型性淋巴组织细胞浸润形成的肺实质肿块和结节,现已证明本病的某些组织学特征对预后有一定影响,在炎性浸润灶内,半数以上浸润细胞为非典型性淋巴网状细胞,提示预后不良,而浸润细胞主要为小淋巴细胞,或者浆细胞大于10%,预后较好,坏死的程度对预后无影响。
神经系统也是本病常见的受累部位,尸解发现脑膜,血管和脑实质有典型性且具有浆细胞样多形细胞广泛浸润,在广泛坏死区内可见有神经细胞丧失和白质碎裂,也可见有脑血管局灶性阻塞伴管壁坏死,管壁呈非典型性淋巴网状细胞浸润,血栓形成,管腔狭窄和动脉瘤形成,部分死亡病例尸解时尚可见脑和脊髓内有广泛性坏死区,可伴有免疫母细胞肉瘤,个别病例尸解时发现中脑和脑桥有广泛坏死,其脑组织由非典型性淋巴组织细胞所取代。
皮损的组织学所见与肺所见相似,可见到轻到重度皮肤血管中心性和血管破坏性血管炎及混合细胞性肉芽肿性浸润,也可见到不典型的淋巴网状细胞,但嗜酸细胞少见,可出现皮肤附属结构如汗腺和神经的继发炎症和破坏,深静脉一旦受累,则可因邻近脂肪组织坏死而导致脂膜炎,从超微结构上看,组织细胞增生表现出一定特点,包括细胞膜边缘波动,高尔基器发育良好以及胞浆内微丝,微管的聚集等,皮下组织受累时,典型改变为局灶性脂肪坏死,并伴有异物反应。
肾脏的组织学改变表现有典型淋巴网状细胞浸润性结节,这种改变类似梗死,并可见有坏死和血管炎,动,静脉闭塞,病变只见于肾实质,而肾小球无改变,肝脏的病理特征表现为在肝实质内可见有散在的局灶性浸润,大块坏死性病变,在坏死区内可见有大量胶原沉着,血管周围有致密的非典型淋巴组织细胞浸润等。
诊断标准
1.诊断标准
(1)发病年龄和性别:本病可见于各年龄阶段,老年相对多见,平均发病年龄为50岁,男女比为1.7∶1。
(2)临床表现:呼吸道症状最多见(80%),可伴发热,消瘦,皮肤病变也较多见(40%~50%),表现为大片浸润性红斑,结节,溃疡等,中枢神经系统也常常受累(30%),可表现为精神异常,共济失调偏瘫,抽搐;脑神经受累可出现Bell麻痹,暂时性失明,复视,突眼,视力下降或眩晕;周围神经受累出现下肢感觉异常,少部分患者有肝脏受损(25%),可有肝,脾大等,但浅表淋巴结极少受累,这一点与淋巴瘤相反。
2.诊断步骤
临床上出现上述肺部病变,或伴随皮肤,神经系统病变,或伴肝,脾,淋巴结肿大时,要考虑到LG的可能,进一步可行胸部X线检查,如出现多发结节影伴空洞形成是重要的诊断线索,但必须经病理证实才能最后确诊,本病肺部病变极为常见,常为患者就诊的主诉,应引起重视。
临床出现上述肺部病变,或伴随皮肤,神经系统病变时,要考虑到LG的可能,胸部X射线检查显示伴空洞形成的多发结节阴影是重要的诊断线索,但必须经病理证实才能最后确诊。
鉴别诊断
本病诊断困难,为确定诊断需要活体组织检查,需与Wegener肉芽肿及转移性肿瘤等下列疾病相鉴别:
1.淋巴瘤
该病也常见血管浸润及广泛坏死,LG患者主要表现为纵隔增大,肺门淋巴结肿大以及肝,脾,浅表淋巴结肿大时需和恶性淋巴瘤鉴别,鉴别还是靠病理,淋巴瘤浸润的细胞较为均一,免疫组化染色检测为单克隆性,而LG主要表现血管坏死性肉芽肿,伴有各种类型的细胞浸润,免疫组化显示淋巴细胞主要为CD4 阳性的T细胞,TCR基因克隆性重排(-),两者显然不同,以下特点可与LG区别:
(1)常伴浅表,肺门,纵隔淋巴结及肝脾肿大。
(2)浸润细胞常为单一种细胞。
(3)免疫过氧化物酶检查为单克隆免疫球蛋白,而LYG为多克隆免疫球蛋白,但LG中如发现灶性单一异型淋巴细胞浸润时应警惕其发展为恶性淋巴瘤的可能。
2.韦格内肉芽肿
(Wegener肉芽肿WG) 与LG相似,肺部受累最为常见,但本病临床上更为少见,无性别差异,以40~50岁多见,患者除有发热,关节痛,肌肉痛,贫血,白细胞增多,血沉增快外,呼吸道症状早期表现为鼻,中耳,鼻窦的炎症及咽喉,支气管的溃疡性损害,在中,末期出现灶性肾小球肾炎,皮肤及黏膜广泛性出血性皮疹,弛张热,多因尿毒症或全身衰竭而死亡,病程平均约6个月,多在3个月以内,十分凶险,此病虽和LG均有血管坏死性肉芽的病理特点,但WG具有特征性的多核巨细胞,肉芽肿常有较多的中性粒细胞及细胞碎屑,而淋巴细胞较疏松,且凝固性坏死少见,另外,血清中抗中性粒细胞抗体(ANCA)阳性是诊断WG的重要依据,其特异性为86%,敏感性为78%,可与LG相鉴别,病理上浸润细胞主要由中性粒细胞,组织细胞及少量嗜酸粒细胞组成,并间杂有稀疏的淋巴细胞和浆细胞群,无淋巴瘤样肉芽肿出现的特征性非典型性淋巴网状细胞浸润明显的增生活跃现象,韦格内肉芽肿在浸润灶内或其附近可见有嗜酸粒细胞,而淋巴瘤样肉芽肿浸润灶无此改变,另外,类上皮样细胞和巨细胞是韦格内肉芽肿常见的组织学特征,但很少见于淋巴瘤样肉芽肿,其他肺血管炎和肉芽肿病LG和Wegener肉芽肿鉴别困难,但后者浸润细胞无异型淋巴细胞。
3.感染性肉芽肿病
在确定LG之前仍需除外分枝杆菌,真菌等感染性疾病所致的肉芽肿病。
4.阻塞性支气管炎
由于肉芽肿病变破坏支气管壁及纤维增生而造成管腔狭窄或闭塞,可误诊为阻塞性支气管炎,必须取肺组织活检以排除阻塞性支气管炎。
5.肺转移性肿瘤
肺转移性肿瘤也常可表现为肺部多发性结节,当尚未发现原发灶时应与LG鉴别,转移性肿瘤的结节常为圆形,边缘清晰,光滑,密度均匀,无空洞,一般无大片浸润影,而LG常表现为伴有空洞的结节形成,可见到肺大片浸润性阴影,病理上两者截然不同,肺转移瘤大多可查到原发灶,如肝,前列腺,乳腺等,伴有原发病灶的症状可资鉴别。
淋巴瘤样肉芽肿西医治疗
本病治疗至今尚无满意的方法。据报道,在病程早期用糖皮质激素治疗,可获得一定疗效。用激素与免疫抑制剂联合治疗,较单用激素治疗效果好。这些药物的用法和剂量与韦格内肉芽肿相同。有人报告本病即使接受激素或免疫抑制剂治疗,也不能阻止其发展。多数学者认为当本病已出现肺内广泛性病变或者中枢神经系统病变时,即使进行充分的化疗,也难取得满意的疗效。这充分说明早期诊断与治疗本病的重要意义。
1982年Fauci用泼尼松(1mg/(kg·d),2个月后减量,持续2年余)及环磷酰胺[2mg/(kg·d),持续达37个月]治疗,使46%病人得到5年左右缓解。局限性病变可行放疗。G1型宜单用肾上腺皮质激素,G2或G3型可选用治疗ML的联合化疗方案,约半数患者可完全缓解。本病中丘疹和皮下结节可自然消退,更多见复发。全身性皮质激素和环磷酰胺治疗皮损有效。对某些难治性皮损可应用放射治疗。
1.内用治疗
皮质类固醇激素适用于早期限局性病变即良性淋巴细胞性血管炎及肉芽肿,采用中量到大剂量的皮质类固醇激素可缓解病情,常用量泼尼松60~80mg/d。亦可应用免疫抑制剂治疗,早期联合应用环磷酰胺和皮质激素可使病情长期缓解。
2.放射治疗
孤立性病灶可行小剂量放射治疗。
预后
本病预后不良,大多在发病后2年死亡,平均存活11.3个月,死亡率达65%~90%,其中50%死于呼吸衰竭。约10%~50%病例发展为恶性淋巴瘤。本病预后与肺和肺外部位受累程度以及组织学分级相关。一般说来,预后与异型大细胞百分比成反比。一般可用由Lipford等设计出评价“血管中心性免疫增殖性损害(AIL)”计分系统来进行组织学分级。一级AIL相当于“良性淋巴细胞血管炎”,对苯丁酸氮芥治疗有效,二和三级AIL代表“血管中心性淋巴瘤”,其中二级AIL相当于“经典LG”或“混合淋巴细胞性和大淋巴细胞性血管中心性淋巴瘤”,三级AIL相当于“大淋巴细胞性血管中心性淋巴瘤”。直到最近,近2/3的二或三级血管中心性淋巴瘤在诊断一年内死亡。化疗的进展对预后略有改善。
多死于肺部病变和中枢神经系统病变。神经系统病变的发生与预后直接相关,一旦神经系统受累,其病死率高达80%以上。若在病程早期用激素和免疫抑制剂治疗,则可使部分患者的症状获得较长期的缓解。有12%病人发展为恶性淋巴瘤。死亡率为60%,通常在出现症状14个月内死于泛发性肺部病变和继发感染。1/4患者可带病生存,中数生存期约2年。临床上肺部双侧病变、伴神经系统损害,病理上以不典型淋巴细胞为主者,预后均差。
淋巴瘤样肉芽肿中医治疗
当前疾病暂无相关疗法。
(以上提供资料及其内容仅供参考,详细需要咨询医生。)
加强营养、禁食生冷油腻,注意温补。
1.维护环境卫生,加强身体锻炼,提高自身免疫功能,防止感染。
2.注意生活规律,劳逸结合,心情舒畅,避免剧烈精神刺激。
3.加强营养,禁食生冷油腻,注意温补。
4.避免感冒受凉,防止继发细菌感染。
5.早期诊断,早期治疗。
1.血常规及血沉 少数患者有严重贫血,白细胞可升高或降低,淋巴细胞可增多,血沉加快。
2.尿常规 一般正常,有时可见有轻度蛋白尿和白细胞。
3.生化学检查 当肝实质广泛受侵犯时,转氨酶可升高。
4.免疫学检查 约半数患者可有IgG或IgM升高,细胞免疫试验多为阴性,类风湿因子,狼疮细胞,抗核抗体均为阴性。
5.外周血检查 可有贫血,白细胞减少或增多,淋巴细胞增高或降低。
6.血液检查 血沉可正常或增快,类风湿因子可阳性,RF常阳性,ANA常阴性。
7.免疫球蛋白检查 免疫球蛋白IgA,IgG可轻度增高。
8.病理检查 主要在真皮深层附属器周围有中性粒细胞浸润,还可见淋巴细胞,浆细胞和组织细胞,偶见异形淋巴样细胞,其核大,深染,形态不规则,可见不典型有丝分裂,典型浸润以血管为中心并破坏血管,若皮下组织受累,可引起脂膜炎和灶性脂肪坏死,出现异物巨细胞反应和多种炎症细胞浸润,血管炎在炎细胞浸润稀疏区更明显,主要侵犯细动脉,血管内皮肿胀,腔内有血栓形成,管壁有纤维蛋白样沉积和炎症细胞浸润,浸润中可见异形淋巴样细胞,浸润区内皮肤附属器常被破坏,肺部浸润由小淋巴细胞,浆细胞以及不等量异形淋巴样细胞组成,肾损害主要为肾间质浸润和血管炎,中枢神经损害也为坏死性血管炎和异形淋巴样细胞浸润,肿块以血管为中心的坏死性肉芽肿,血管被多种细胞浸润,包括小淋巴细胞,组织细胞,免疫母细胞,浆细胞等,主要累及中等大小动静脉,血管内膜增厚,管腔狭窄或闭塞,可有血栓形成,根据浸润细胞的类型及坏死范围可分为3级,增生的淋巴细胞主要是T细胞,为CD4 的T细胞,TCR基因重排常阴性,淋巴结活检大多示反应性增生。
9.免疫表型 EBV阳性,B细胞常表达CD20和CD79a,CD15阴性;LMP1阳性,部分单克隆细胞,胞质Ig阳性,不典型淋巴细胞可CD3阳性,其中CD4 细胞为多。
10.X线检查 X线胸片所见依病变进展时期而不同,典型者表现示两肺中,下叶有多发性结节状阴影,大小不一,直径自数毫米至10cm不等,1/3伴有厚壁空洞形成,20%左右仅为单侧肺结节阴影,少数表现为肺大片浸润性阴影,1/3可见胸膜腔积液,但肺门淋巴结不肿大,偶尔可见两肺呈弥漫性网状结节性和绒毛状肺泡浸润,或呈多发性结节性病变,类似于转移性肺癌,病变多为双侧性,主要累及两下肺野,特别是两肺外带,结节影可迅速增大或缩小,而且甚至可以完全消失,少数患者有纵隔或肺门淋巴结肿大。
11.根据病情,临床表现,症状,体征,选择做心电图,B超,CT,MRI等检查。
最常见的并发症为呼吸困难,尤其见于纵隔或肺的淋巴结肿大者,临床病情与淋巴瘤相似的患者,约1/3可出现肺空洞,可因大咯血而死亡,也可发生肺叶支气管部分阻塞,肺实质大片破坏引起呼吸衰竭也是本病的主要死亡原因,中枢神经系统病变可累及脑和脊髓的任何部位,可出现失语,轻偏瘫,失明,运动失调,截瘫,动眼神经麻痹,脑神经麻痹,头痛,感觉异常,意识模糊,昏迷,抽风,四肢瘫,视神经水肿,耳聋,面瘫,感觉迟钝,脑水肿,脑膜脑炎等,广泛浸润肝脏,可造成进行性肝功能衰竭而死亡,尸解发现肝,脾,淋巴结,肾,心脏和胃肠道均可受侵犯。