α1抗胰蛋白酶缺乏性肝病(alpha 1 anti-trypsin deficiency-associated liver disease)是遗传性α1抗胰蛋白酶缺乏引起的代谢性肝脏疾病,以常染色体隐性方式(autosomal recessive fashion)遗传伴等显性表达(codominant expression)。
α1-AT缺乏性肝病可在婴幼儿期初次被发现,也可在此期无肝脏病变表现,到成年后出现慢性肝病表现,8%~12%的PiZZ型α1抗胰蛋白酶缺乏的新生儿在出生后1个月内即发生胆汁淤积性黄疸,血清胆红素可高达340 µmol/L,血清碱性磷酸酶(ALP)活性可达150~1300U/L,患儿体重增加缓慢,嗜睡,易激怒,出现无胆汁粪便,3个月月龄的小儿半数出现高转氨酶血症,血清谷草转氨酶(AST)活性可达80~600U/L,12%~15%的α1抗胰蛋白酶缺乏患儿出现肝硬化病症,表现为腹胀,肝脾肿大,因食管静脉曲张引起的上消化道出血,也可有紫癜等其他部位出血倾向,大多数患者新生儿肝脏淤胆持续7个月左右至1年可消退,如无好转,则可逐渐发展出现进行性肝损害,进展为肝硬化甚至死亡,引起的肝硬化也可在成年时发生,但在中老年期才出现肝硬化者很少,成年期发病多见于杂合子型α1-AT缺乏性肝病患者,病情发展较缓慢,临床表现各异,有报道成人杂合子型α1-AT缺乏性肝病患者肝功能衰竭的危险性明显增加,Sreger报告的120名PiZZ型新生儿中有14名长期有梗阻性黄疸,新生儿期肝炎和幼年期肝硬化,成年期肺气肿,成人α1抗胰蛋白酶缺乏肝硬化可无症状,可出现肝硬化的临床表现,也可伴发肝癌,多见于50岁以上的纯合子型患者。
对于任何非感染性慢性肝炎患者,原因未明的肝脾肿大,肝硬化和门静脉高压患者都应考虑有α1-AT缺乏性肝病的可能性,也应警惕少数肝癌是因α1-AT缺乏引起。
血清蛋白电泳可见α1球蛋白缺乏常提示该病,直接测定α1抗胰蛋白酶可确诊,但应重视遗传表型分析,因为α1-AT的产生受这些因素影响,所以诊断应根据表型分析,而不单根据α1-AT水平检测。
肝细胞活检。
(一)发病原因
α1-抗胰蛋白酶(α1-AT)缺乏性肝病是血清中一种拮抗蛋白酶的成分α1-抗胰蛋白酶缺乏而引起的一种先天性代谢病。
(二)发病机制
在全身细胞中产生的血清蛋白酶可引起靶器官的炎症性连锁反应和补体活化,正常人体内也存在一组抑制蛋白酶活性的物质,称为蛋白酶抑制物(proteinase inhibitor),广泛分布于血浆,淋巴液,尿液,唾液,泪液,支气管分泌物,脑脊液,宫颈黏液,精液,初乳等体液和一些组织细胞的胞质中,蛋白酶抑制物参与多种生理和病理过程,α1抗胰蛋白酶(α1-AT)为血清中主要的蛋白酶抑制物,系由肝细胞合成的一种糖蛋白,分子量50000~60000,在电泳分离时处于α1的位置,释入血浆后,构成α1-球蛋白的主要成分,约占α1-球蛋白的90%,全部血清蛋白的4%,α1-AT是一种急性时相反应的蛋白酶抑制剂,可以抑制多种内源性和外源性蛋白酶,如胰蛋白酶,糜蛋白酶,弹性蛋白酶,凝血酶,纤溶酶,血管舒缓素以及某些细菌和病毒具有蛋白酶性质的产物等,还具有中和毒素,清除毒素,控制感染,炎症,阻止自身消化等多种功能,在有炎症反应,应激,创伤,妊娠或肿瘤时也可刺激α1-AT释放,血清该酶的水平可升高。
α1-AT与弹性蛋白酶形成1∶1的牢固的复合体,以后在血液循环中被分解,这种复合体可与肝细胞的血清蛋白酶受体结合,从而刺激肝细胞产生α1-AT,在正常情况下,α1-AT可控制90%以上的血清弹性蛋白酶的活性,α1-AT分子上的唾液酸(N-乙酰神经氨酸)对其生物活性非常重要,祛除唾液酸的残基α1-AT即失去活性,并且很快从血液循环中清除,不含唾液酸残基的α1-AT在肝细胞合成后不能被释放入血,蓄积在肝细胞,可引起肝细胞损伤。
迄今有关α1-AT缺乏的家系研究报告表明,α1-AT等位基因(alleles)呈等显性表达蛋白酶抑制物基因(Pi基因),Fagerhol等认为,控制α1-AT合成的所谓Pi基因是位于常染色体上的等位基因,采用薄层凝胶聚焦技术分析人类α1-AT电泳迁移率,发现其在人群中存在多态现象,现已鉴定出75种以上的α1-AT变异体,但大多数无临床意义或很罕见,分别命名为B,C,D,E,F,G,L,M,N,P,S,V,W,X,Z等,各等位基因分别用PiM,PiS,PiZ等表示,纯合子的基因型用PiMM,PiSS等表示,杂合子用PiMZ,PiSZ等表示,以上统称为Pi基因系统,编码α1-AT的基因定位于14号染色体长臂(14q24.3-32.1),Pi基因系统的各种表现型,其血清蛋白酶抑制活性与α1-AT的浓度是不同的,PiM是具有正常功能的基因,绝大多数正常人是PiM的纯合子(PiMM),其血清中α1-AT含量正常,功能也正常,具有PiZ基因的纯合子(PiZZ)个体血清中α1-AT含量严重缺乏,仅为正常人的15%左右,这种人常发生阻塞性肺病和幼年型肝硬化,具有纯合子PiSS血清中α1-AT含量中度缺乏,约为正常人的60%,这种人亦有患肺气肿和肝硬化的倾向,杂合子PiMZ,PiSZ等个体也有发生肺气肿和肝硬化的倾向,Jeppson等分析肽图(peptid mapping)发现α1-AT缺乏症PiZZ变异型蛋白肽链中一个谷氨酸被赖氨酸所取代,一个谷氨酸被谷氨酰胺所取代,PiSS变异型系谷氨酸被缬氨酸所取代。
α1-AT在肝细胞的粗面内质网产生,转运到Golgi器供分泌之用,有一种假说,即与等位基因突变有关的蛋白错误折叠(misfolding)构象,可能使α1-AT滞留在内质网而不能释放到Golgi器,因为有此错误折叠的变化,正常的隐蔽区可能暴露,从而与不同的配体受体接触,而不能作为有效的分子释放,异常的α1-AT滞留在内质网造成蓄积,排泌减少,其在细胞内的降解率取决于基因调控,α1-AT缺乏引起肝细胞损害的病理生理尚有争议,目前认为肝损害使继发于α1-AT在肝细胞粗面内质网的蓄积并有可能改变异常的α1-AT在肝细胞内的降解,α1-AT缺乏症患者的纯合子和杂合子的肝细胞内质网内,可见过碘酸Schiff试验(periodic acid schiff,PAS)阳性的耐淀粉酶颗粒,支持这一假说。
α1-AT缺乏者有3个转归:一部分人可能终身健康;大部分人在青中年期患严重的肺气肿;一部分人在婴儿期就已患肝脏疾病,但是很少有同时患肺气肿和肝硬化者,目前尚不清楚为何一些人发生严重的肝病,而另一些人却无症状,多认为肝损害是由多方面因素引起的,如弹性蛋白酶能分解弹力纤维造成肺气肿病变,但在正常情况下,弹性蛋白酶抑制因子可抑制此酶的活力,避免肺气肿,研究发现PiZ较易发生慢性阻塞性肺疾病(COPD),先天性α1-AT缺乏具遗传易感性,需与后天外界因素结合才会产生致病作用,吸烟具有更大的危险性,如吸烟者肺巨噬细胞增多,细胞溶酶体多而大,烟草燃烧产生的NO2可刺激肺内巨噬细胞及中性粒细胞释放弹性蛋白酶,而α1-AT缺乏者由于抑制蛋白酶的能力减弱,易发生肺组织损伤,从而引起慢性阻塞性肺疾病,α1-AT缺乏发生肝硬化者与肺部疾病无关,α1-AT缺乏是α1-AT缺乏性肝病的主要因素,还有其他因素参与,体内蛋白酶活性增高,是肝脏对其他一些致病因素和有毒物质的易感性增加,致使肝损害,Gam提出也有可能由于肠屏障破坏或有缺陷,肠内的毒素被吸收入肝,由肝Kupffer细胞摄取释放出溶酶体酶,当人体缺乏α1-AT时该酶具有破坏性;或由于肝细胞内α1-AT滞留,肠毒素进入肝脏后,肝细胞内具有保护作用的蛋白溶解酶被过多的α1-AT抑制而使肝细胞受损;或因肝细胞内α1-AT过多而抑制了肝脏内源性蛋白酶的产生,以至不能对抗肠源性有毒物质,从而引起肝脏的损害。
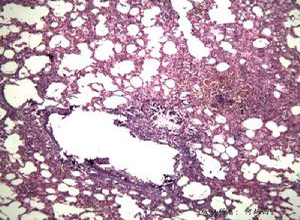
病理组织学改变因患者年龄而异,患病婴儿的肝活标本检查显示胆管缺乏(bile duct paucity),肝细胞内胆汁淤积,伴有或不伴有细胞肿大变形,轻度炎性改变或脂肪变,肝细胞内可见一些特征性的PAS阳性的耐淀粉酶样小体(diastase resistant globule),这种小体能被荧光素标记的α1-AT抗血清强烈染色,具有α1-AT抗原性(图1),这种颗粒状的包涵物位于肝细胞内质网上,随着年龄的增长而增多,说明患者α1-AT的缺乏是由于合成的α1-AT不能释放入血而蓄积于肝细胞内,以纯合子PiZZ表现型患者为多,婴儿期α1-AT缺乏的肝病患者,如无好转可发生进行性肝损害,在门静脉区纤维组织明显增生,逐渐形成小叶间纤维化,可进一步呈小结节型或大结节型肝硬化,纯合子型α1-AT缺乏可引发原发性肝癌。
1.淤胆型药物性肝损害与肝外阻塞性黄疸的鉴别有赖于详细询问病史,各种过敏表现及B超,CT,MRI,逆行胰胆管造影等检查而鉴别。
2.肝硬化有慢性肝炎,血吸虫感染,长期酗酒等病史,肝功能,B超,CT,肝脏活体组织检查等有助于鉴别。
α1抗胰蛋白酶缺乏性肝病西医治疗
治疗
本病尚无有效的特殊治疗方法,基本按肝硬化的一般治疗方案进行治疗。改善症状和适当的营养支持。为减轻胆汁淤积性肝病病情,患病婴儿应尽可能给予母乳喂养,给予脂溶性维生素,可试用熊去氧胆酸治疗。要戒烟和杜绝被动吸烟。
曾有报道应用巴比妥类药物、皮质激素、免疫抑制药治疗本病,但均无效果。
α1抗胰蛋白酶增补治疗(augmentation therapy):旨在增加肝脏内源性α1抗胰蛋白酶释放,从而增加抗弹性蛋白酶的活性,像减轻肺损害一样来达到减轻肝损害。但这种方法也同时增加了α1抗胰蛋白酶与血清蛋白酶复合体受体的结合,会刺激异常的α1抗胰蛋白酶产物的增加,导致其在肝细胞内的蓄积,从而加重对肝脏的损害。所以这种方法不适合治疗α1抗胰蛋白酶缺乏性肝病。
肝移植已被用于治疗进展到晚期的α1抗胰蛋白酶缺乏性肝病的患者,本病是适合肝移植治疗的最常见的代谢性肝病之一。肝移植除了替代已有损害的肝脏外,还可矫正代谢缺陷,以免进展到全身性病变。
α1抗胰蛋白酶缺乏性肝病被认为是可通过基因治疗来重建正常的基因型的许多疾病之一。其潜在的益处是可减少对于肝移植的需求。α1抗胰蛋白酶缺乏性肝病基因治疗是通过对α1-AT缺乏的肝细胞的基因组内加进正常的α1-AT基因,使细胞能合成正常的α1-AT。此外,其他一些治疗方法业已进行研究。如研制血清蛋白酶复合体受体阻滞药(serping-enzyme complex receptor blocker)用于减少异常的α1-AT的产生,或阻断内质网α1-AT结合位点,以避免有效地分泌异常的α1-AT。这是治疗研究的前沿。
α1抗胰蛋白酶缺乏性肝病中医治疗
暂无可参考数据。
α1抗胰蛋白酶缺乏性肝病日常保健
配合药物食用富含脂溶性维生素的食物。
维生素A是由胡萝卜素转变而成的。胡萝卜、油菜、菠菜、韭菜中含胡萝卜素很多,动物性食物中以鸡肝、鱼肝含量最高,猪肝、蛋类含量稍少一些,市面所卖的浓缩鱼肝油,含量最为丰富,每10毫升含维生素A50万国际单位。
维生素D可分为两种,维生素D2和维生素D3。
维生素D3主要是由人体自身合成的,人体的皮肤含有一种胆固醇,经阳光照射后,就变成了维生素D3。还可来自动物性食物,如肝类,尤其是由海产类的鱼肝中提炼的鱼肝油含量最丰富。
维生素D2来源于植物性食物,酵母、蕈类等含量较多。
维生素E广泛存在于动植物食品中,以植物油含量最多,但维生素E不稳定,储存及烹调过程中均会有损失。
维生素K,食物中如肝类、蛋类、豆类中都有。
α1抗胰蛋白酶缺乏性肝病日常预防
1.禁烟 α1-抗胰蛋白酶缺乏者应绝对禁烟,吸烟可加重α1-抗胰蛋白酶缺乏性肺气肿。
2.防治并发症 对只有轻微肝损害者只需长期支持治疗;对有门脉高压者可行门腔或脾肾分流术。
3.肝移植 对肝损害严重者应行肝移植,由于肝是合成α1-抗胰蛋白酶的惟一场所,因此肝移植不仅能治愈肝病,且能纠正α1-抗胰蛋白酶缺乏,现认为肝移植是治疗Pizz终末期肝硬化的有效方法,应用PiMM表型的供者肝脏做肝移植,可望提高其存活率及改善病情。
1.测定血清α1-抗胰蛋白酶 浓度(正常值2000~3000mg/L):比正常减少10%~15%,对诊断可能有帮助,但不能确诊,因在急性炎症时,血清α1-抗胰蛋白酶浓度可能增加。
2.pi表型分析 应用等电聚焦或酸性条件下琼脂电泳鉴定α1-抗胰蛋白酶表型可建立诊断,目前,PCR技术已用于检测α1-抗胰蛋白酶变异体,此法不仅迅速,敏感性高,而且只需极少量的细胞物质,该技术对确定诊断,人群筛检及出生前诊断等均有用。
肝穿刺活组织检查:显示肝硬化,PAS染色可见肝细胞内特征性包涵体,荧光染色显示在肝细胞内蓄积有蓝色颗粒,即α1-抗胰蛋白酶抗体荧光带。
肝脾肿大,因食管静脉曲张引起的上消化道出血,新生儿肝脏淤胆如无好转,则可逐渐发展出现进行性肝损害,进展为肝硬化甚至死亡。