先天性颅锁骨发育不全是一种先天性全身性膜性骨骨化不全,以颅顶骨与锁骨发育障碍为主要特征,可多骨或单骨受累。近年来的研究支持发病的免疫学机制,有多种免疫学理论其他的病因包括中毒,微量元素、氨基酸代谢障碍病毒感染。部分病例有遗传特征。关岛地区发病率高出世界其他地区50~100倍但未探明环境毒物,饮食结构异常的证据。
病变累及面较广,除主要累及锁骨,颅骨以外,尚可存在骨盆骨化不完全,脊柱畸形变扁,附件缺如或隐性脊柱裂等,牙齿以及腕骨,骨钙化缓慢等情况,症状表现主要为头大面小,肩下沉,狭胸,前客,头顶及下颌相对较大,锁骨可部分或完全缺如,通常为对称性,有时合并有肌肉异常,如三角肌的前部和斜方肌的锁骨部缺如。
本病病因不明,为常染色体显性遗传,其侯选基因目前已被定位到6P21 ,大约2/ 3 有家族史,1/ 3 为散发,性别间无明显差异,该病可在任何年龄发病,但在出生后2 年内发病最显著。
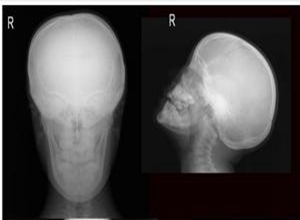
头面部的典型畸形及X线片所见,很容易做出诊断。 X线检查方法为确诊本病的主要手段,由于该病X线征象具有以上特殊性,所以诊断并不困难,关键在于能想到本病,必要时仍需与以下疾病相鉴别:
(1) 佝偻病:佝偻病所显示的方颅,囟门闭合延迟锥体及骨盆变形有时与该病相似,但前者无锁骨发育障碍,且经抗佝偻病治疗迅速好转。
(2) 软骨发育不全:为全身对称性软骨发育障碍骨骼纵向生长缓慢,而横向生长正常,故管状骨粗短,病人身材矮小且四肢短小,但膜化骨不受累。
(3) 克汀病:虽也显示为颅骨发育滞迟,颅缝增宽,并见缝间骨,但无锁骨,坐骨,耻骨的缺损及骨化中心延迟等,患儿明显智力低下。
(4) 成骨不全:由于膜内化骨作用低下,有时也见囟门及颅缝闭合延迟,缝间骨等,但其主要表现为骨脆易折及骨痂形成过盛,有时伴有不同程度蓝巩膜,听力障碍等。
(5) 与外伤有关的锁骨部分缺如畸形等,此种情况经询问病史将不难发现。
先天性颅锁骨发育不全西医治疗
本病临床症状相对较少,典型病例诊断较为明确,虽畸形复杂,外貌丑陋,但多数患儿智力正常,生活及劳动不受影响,很少引起严重功能障碍,不需作特殊治疗,除非合并肢体畸形而影响活动时或偶因锁骨残端压迫肱动脉或臂丛神经时,予手术切除锁骨残端解除压迫,效果满意。
先天性颅锁骨发育不全中医治疗
当前疾病暂无相关疗法。 (仅供参考,详细请询问医生)
1、饮食上应注意清淡,多以菜粥、面条汤等容易消化吸收的食物为佳。
2、可多食新鲜的水果和蔬菜,以保证维生素的摄入量。 3、给予流质或半流质的食物,如各种粥类、米汤等。本病为先天性疾病,故无有效预防措施,早诊断早治疗是本病的防治关键,诊断时还应注意与其它具有相似症状的疾病进行鉴别,以便给患儿正确的治疗。
本病主要是进行X线检查,有学者按有无遗传学关系及症状的轻重不同分为三类:
(1)第一类为标准型::有家族遗传关系,颅骨,锁骨,骨盆等均受累,其X 线表现典型,如单侧或双侧锁骨部分或全部缺如,颅骨骨化不全,前囟门大,颅骨缝未闭,有时胸骨柄缺如,广泛脊柱裂等。
(2)第二类为家族遗传型:有家族遗传关系,但颅骨不受累。
(3)第三类为散发型:无家族遗传关系。
本病同时也会合并软骨发育障碍,严重者表现为侏儒、耻骨发育障碍,脊柱裂及腕骨发育滞迟等,有时还会合并有肌肉异常,如三角肌的前部和斜方肌的锁骨部缺如。