糖原贮积病Ⅶ型(GSD-Ⅶ)是由于肌磷酸果糖激酶缺陷所造成,较罕见,以肌肉组织受损为主。糖原贮积病(glycogen storage disease,GSD)是一类先天性酶缺陷所造成的糖原代谢障碍疾病。这类疾病的共同生化特征是糖原储存异常,多数病种是糖原在肝脏、肌肉、肾脏等组织中储积量增加。
临床表现主要为肌肉无力,以体能活动能力降低和肌疼痛性痉挛为特征,需要体能较大的剧烈运动,如提携重物,快跑,上楼或攀登等均可造成肌痛,肌痉挛和肌僵硬;休息或减慢活动速度即可使症状缓解,症状严重与否与运动量大小和时间的长短成正比,一般多发生在四肢肌痛,无低血糖发作。
1.典型表现 有以下5点特征:
(1)本型通常在儿童期即出现运动耐量减低,且较Ⅴ型患者为重,可伴有恶心,呕吐,在剧烈运动后常出现肌肉痛性痉挛和肌球蛋白尿。
(2)伴有溶血特征,血清胆红素增高,网织红细胞数增高。
(3)高尿酸血症常见,且在运动后较GSD-Ⅴ或Ⅲ型患者增高更为明显。
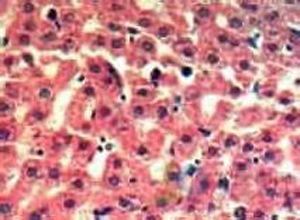
(4)肌活检可显示肌纤维中有类似支键淀粉的异常糖原累积,Schiff试验阳性,但不能被淀粉酶水解。
(5)在进食富含碳水化合物的饮食后,对运动特别不能耐受,这是因为本型患儿的肌肉系统不能利用食物中的葡萄糖,而血中高浓度的葡萄糖却抑制脂肪分解,致使肌细胞中的脂肪酸和酮体迅速被耗尽所致。
2.变异型表现 除上述典型患儿外,另有较罕见的2种变异型:
(1)成人型:婴儿期即发病,患儿肌张力低下,肢体肌力差,表现为进行性肌病,常在4岁左右夭折。
(2)成人型:以慢性进行性肌乏力为特征,肌肉痛性痉挛和肌红蛋白尿罕见。
(一)发病原因
本型是由于肌磷酸果糖激酶缺陷所造成。
(二)发病机制
磷酸果糖激酶是一个糖酵解途径中主要的酶,其作用是催化6-磷酸果糖转换成1,6-二磷酸果糖,该酶由3个同工酶亚单位组成(M-肌肉,L-肝脏,P-血小板);不同的同工酶由各自的编码基因在不同的组织中表达,骨骼肌中含有的是M亚单位,其编码基因位于lcen-1q32,基因突变即造成肌肉中酶的完全缺乏和红细胞中该酶活力下降约50%(细胞中含M和L的杂交型酶)。
根据病史,体征和血生化检测结果可做出临床诊断,病儿白细胞及肌组织的磷酸果糖激酶活力明显降低可助诊断。
与糖原贮积病Ⅴ型鉴别。
小儿糖原贮积病Ⅶ型西医治疗
各型糖原累积病都是由糖原合成或糖原分解过程中某种酶缺如或活性减低。这些酶缺陷与酶的相关基因发生突变有关,对于此种基因突变性疾病,目前医学界尚无特殊治疗方法。患者应避免剧烈运动以防止肌痉挛和肌球蛋白尿症。
预后
本病预后较糖原贮积病Ⅴ型差,可发生严重并发症造成死亡。
小儿糖原贮积病Ⅶ型中医治疗
暂无可参考资料。
以上提供资料及其内容仅供参考,详细需要咨询医生。
日常保健
食物品种多元化,充分发挥食物间营养物质的互补作用。
患者应避免剧烈运动以防止肌痉挛和肌球蛋白尿症。
实验室检查可见患者血清肌酸激酶水平增高,运动后更甚;由于在运动时,供应肌肉能量的ATP不足,因此嘌呤核苷酸代谢旺盛,以致血中氨,肌酐,次黄嘌呤和尿酸等浓度亦上升;高尿酸血症常见,且在运动后较GSD-Ⅴ或Ⅲ型患者增高更为明显。
剧烈运动后可有肌球蛋白尿,伴有溶血特征,血清胆红素增高,网织红细胞数增高,溶血性贫血。
肌活检可显示肌纤维中有类似支键淀粉的异常糖原累积,Schiff试验阳性但不能被淀粉酶水解,对活检肌组织进行酶学或组织学检测可提供确诊依据;也可用血细胞或成纤维细胞检测M型磷酸果糖激酶同工酶活力。
常规做X线,心电图,B超和肌电图检查,表现为进行性肌病改变。
可发生肌球蛋白尿,伴有溶血血清胆红素增高,溶血性贫血,高尿酸血症,进行性肌病等。