糖原贮积病Ⅵ(GSD-Ⅵ)系因肝磷酸化酶缺陷所造成,较罕见。患儿多在幼儿期即呈现肝大和生长迟缓,无心脏和骨骼肌受累症状;低糖血症,高脂血症和酮体增高程度均较轻。随着年龄增长,肝大和生长滞后情况也逐渐好转,且常在青春发育期消失。多数患儿无须治疗,为防止发生低血糖,可采取多次少量方式进餐,或给予高碳水化合物膳食。
新生儿期或婴幼儿期均无发病,无明显性别差异,临床上与糖原贮积病Ⅰ,Ⅲ型相似,但较Ⅰ型为轻,临床表现为肝脏肿大,可有轻度血脂及转氨酶增高,可有中度生长迟缓,低血糖少见,由于症状有时轻微可致漏诊,因而有人认为其属良性肝大,无心脏和骨骼肌受累症状,智力正常。
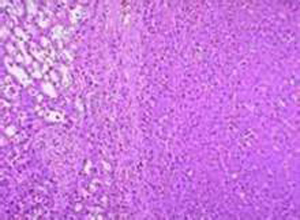
除了上述临床表现外,确诊有赖于实验室结果,患者白细胞中,可测出磷酸化酶减少,活力降低,或肝活组织检查见糖原沉积。
(一)发病原因
本病征因肝磷酸化酶缺陷所致,多数为部分缺陷。
(二)发病机制
糖原是由葡萄糖构成的高分子多糖,主要贮存在肝脏和肌肉中作为备用能量,正常肝和肌组织中分别含有约4%和2%糖原,摄入体内的葡萄糖在葡萄糖激酶,葡糖磷酸变位酶和尿苷二磷酸葡糖焦磷酸化酶的催化下形成尿苷二磷酸葡萄糖(UD-PG),然后由糖原合成酶将UDPG提供的葡萄糖分子以α-1,4-糖苷键连接成一个长链;每隔3~5个葡萄糖残基由分枝酶将1,4位连接的葡萄糖转移成1,6位,形成分支,如是扩展,最终构成树状结构的大分子,糖原的分子量高达数百万以上,其最外层的葡萄糖直链较长,大多为10~15个葡萄糖单位。
糖原的分解主要由磷酸化酶催化,从糖原分子中释出1-磷酸葡萄糖,但磷酸化酶的作用仅限于1,4糖苷键,并且当分支点前仅存4个葡萄糖残基时就必须由脱支酶(淀粉-1,6-葡糖苷酶,amylo-1,6-glucosidase)将其中的3个残基转移至其他直链以保证磷酸化酶的作用继续进行,与此同时,脱支酶可以解除α-1,6-糖苷键连接的一个葡萄糖分子,这样反复进行便保证了机体对葡萄糖的需求,存在于溶酶体中的α-1,4-葡萄糖苷酶(酸性麦芽糖酶)也能水解不同长度的葡萄糖直链,使之成为麦芽糖等低聚糖分子,上述糖原合成和分解过程中任一酶的缺陷即导致不同临床表现的各型糖原贮积病。本病因肝磷酸化酶部分缺陷所致,因此,病变相对较轻。
临床表现特点和实验室,辅助检查特点有助于本症与GSD-Ⅰ,GSD-Ⅲ型相鉴别,胰高血糖素试验显示血糖不增高,依此可发现与GSD-Ⅸ型相区别,后者糖耐量曲线正常。
小儿糖原贮积病Ⅵ型西医治疗
治疗上可使用高蛋白饮食,少量多次。亦可以补充新缺乏的酶(磷酸化酶是催化糖原还原性末端葡萄糖残基的d-1,4-糖苷键断裂,反应,生成1-磷酸葡萄糖和少了一个葡萄糖基的糖原分子,但磷酸化酶的作用仅限于1,4糖苷键),进行替代治疗。多数患儿无须治疗,为防止发生低血糖,可采取多次少量方式进餐,或给予高碳水化合物膳食。
预后
随着年龄增长,肝大和生长滞后情况也逐渐好转,且常在青春发育期消失。预后较好。
小儿糖原贮积病Ⅵ型中医治疗
暂无可参考资料。
以上提供资料及其内容仅供参考,详细需要咨询医生。
日常保健
多摄入一些高纤维素以及新鲜的蔬菜和水果,营养均衡,包括蛋白质、糖、脂肪、维生素、微量元素和膳食纤维等必需的营养素,荤素搭配。
糖原贮积病Ⅵ的预防可参照糖原贮积病的预防方法,应包括妊娠期间预防感染,避免高龄生育,近亲婚配,避免辐射,接触化学物质,遗传物质异常等,预防性优生措施:
1、禁止近亲结婚。
2、婚前检查以期发现不应结婚的遗传病或其他疾病。
3、携带者的检出 通过群体普查,家系调查及系谱分析,实验室检查等手段确定是否为遗传病,并确定遗传方式等。
4、遗传咨询。
5、产前诊断 产前诊断或宫内诊断,是预防性优生学的一项重要措施。
1.生化异常 包括低血糖,酮症酸中毒,乳酸血症及高脂血症,但程度较轻。
2.糖耐量试验 呈现典型的糖尿病特征。
3.肾上腺素试验 皮下注射1∶1000肾上腺素0.02ml/kg,注射前及注射后10,20,30,40,50,60min,分别测血糖,正常者血糖上升40%~60%;糖原累积病患者血糖无明显上升。
4.胰高血糖素试验 肌注胰高血糖素30µg/kg(最大量1mg),于注射后0,15,30,45,60,90,120min分别取血测血糖,正常时15~45min内血糖升高1.5~2.8mmol/L,本症在空腹或餐后无明显血糖升高。
5.肝活检 确诊最好做肝组织活体检查,糖原染色见糖原增多,特异性酶活性降低。
6.基因检测 可通过外周血白细胞DNA分析进行基因检测。
常规做X线,B超和心电图检查,可发现肝大,余无异常。
可有轻度血脂及转氨酶增高,中度生长迟缓。