朗格汉斯细胞的组织细胞增多症(langerhans’cell histiocytosis,LCH)又名朗格汉斯细胞肉芽肿(Langerhan-cell granulomatosis)为一种朗格汉斯细胞增生性疾病,侵犯皮肤和皮肤外的器官。本病可累及皮肤、骨骼,肺、神经系统和其他器官。临床上可分3型:即Letterer-siwe病,Hand-schüller-christian病和嗜酸性粒细胞肉芽肿。但三者之间无明显界限,常可重叠或互相转变,一般而言,如果在一岁内发病,则常以致死性内脏损害为特点,多为Letterer-siwe病,如在幼年发病,常以多发性骨损害为主,而内脏损害轻者,即Hand-schüller-christian病;在较大的儿童和成人,本病通常局限,常表现为1个或数个骨损害,即嗜酸性粒细胞肉芽肿。
-
挂什么科:内科 血液科
-
需做检查:干扰素 浆细胞 皮损
-
治疗方法:手术治疗 药物治疗 放射治疗 支持治疗
-
常用药物:暂无相关信息
-
一般费用:根据不同医院,收费标准不一致,市三甲医院约(10000 ——50000元)
-
传染性:无传染性
-
治愈周期:3个月
-
治愈率:40-60%
-
患病比例:本病罕见,发生率约为0.0001%--0.0002%
-
好发人群:无特殊人群
-
相关症状:
-
相关疾病:
1.Letterer-Siwe病(LSD)
为急性播散性朗格汉斯组织细胞增生症,损害广泛,多脏器受累,全身症状加重,婴儿期发病,1/3病例在出生后6个月内发病,2/3中的大多数亦在2岁内发病,偶可见于较大儿童及成人,80%患者发生皮肤损害,常为本病首发症状,有诊断价值,典型皮损为小而半透明丘疹,皮损广泛而密集,直径1~2mm,略高起,颜色为淡褐色,表面被覆鳞痂,常伴发瘀点,好发于头部,躯干和皱褶部(耳后,腋窝和阴部);发生于头部者,皮损倾向融合,呈脂溢性皮炎样外观;发生于皱褶部者,类似擦烂,黏膜可见糜烂和溃疡,但罕见,此外尚可见甲沟炎,甲皱襞破坏,甲剥离,甲下角化过度和甲床紫癜纹,以上甲症状表明预后良好,出现紫癜则提示预后不良,本病系统性症状常见者为肺受累,见于1/2以上的患儿,可有呼吸困难,发绀,气胸,肺脏可显示典型蜂巢外观;肺损害症状不一,轻者可无症状,重者可以致死,肝肿大为常见系统性损害,严重者特别是伴发黄疸者,则预后不良。
脾大不常见,25%~75%的致死病例,可见淋巴结肿大,60%患者发生骨损害,常见于扁骨,椎骨和颅骨,溶骨性损害可无症状或伴发疼痛和功能性损伤,通常呈多发性,并逐渐出现,慢性中耳炎常见,造血系统受累罕见,如发生自发性血小板减少症和严重贫血则常致死亡,患者常有发热,病情发展迅速,总的来说预后不良,但偶亦见仅有皮损而无系统症状者,则预后良好。
2.Hand-schüller-christian病(HSCD)
为慢性播散性朗格汉斯组织细胞增生症,70%的病例发生于生后2~7岁,91%患者发生于30岁前,典型系统性症状为尿崩病,突眼和骨损害,但其中任何1个或全部症状均可缺无,其中骨损害最常见,约有80%病例发生,好发于颅骨,特别是颞顶骨,损害呈局限性溶骨区,呈典型地图状外观,下颌骨溶骨性损害亦常见,乳突区骨损害可引起中耳炎,尿崩症见于50%以上的病例,常见于颅骨和眶骨受累的患者,突眼症发生较晚,见于10%~30%的患者呈单侧或双侧,少于20%患者可见肺浸润;肝肿大和淋巴结肿大罕见,约1/3患者出现皮肤损害,皮损分3型,最常见者为浸润性斑块,可形成溃疡,特别是腋窝,腹股沟,口腔,阴唇,其次为像Letterer-siwe病所见的广泛性有融合趋势的鳞屑或结痂性丘疹,第3型皮损罕见,呈散在柔软的淡黄色丘疹性黄色瘤,后者在临床和组织病理上均与幼年性黄色肉芽肿不易区别,需借助免疫组化及电镜检查加以区分,本病病程常长达数年,不经治疗约30%患者可致死亡。
3.嗜酸粒细胞肉芽肿(eosinophilic granuloma)
为慢性局限性朗格汉斯组织细胞增生症,为朗格汉斯细胞增生症中的最良型,多见于5~30岁的男性患者,骨肉芽肿损害常呈单发性,好发于颅骨穹隆,肋骨,脊柱,骨盆,肩胛骨和长骨,除非发生自发性骨折和(或)中耳炎损害,骨肉芽肿损害不易被发现,如损害出现症状时,可有局限性疼痛,压痛和软组织肿胀,皮肤黏膜损害罕见,可分2型。1型为像在Letterer-siwe病和Hand-schüller-christian病中所见的广泛性结痂性丘疹或为1至数个像在Hand-schüller-christian病中所见的红色浸润性斑块,倾向形成溃疡;2型皮损可同时出现,本病呈慢性经过,有自愈倾向,在罕见情况下,嗜酸性粒细胞肉芽肿可发展为Hand-Schüller-christian病。
结合免疫组织化学检查来进行分型鉴别。
(一)发病原因
病因尚不明,尽管不少研究考虑其发病与病毒,免疫学和新生物有关,但迄今尚无令人信服的证据,长期以来不少学者怀疑病毒为潜在的病原因子,认为Langerhans细胞异常增生为病毒感染所引起的异常反应,Lahey等在30名LCH患者中的14名(47%)患者损害中查到人类疱疹病毒-6DNA(HHV-6DNA),提出该病毒与LCH的发病有关,Mcclain等则与此相反,未发现腺病毒,巨细胞病毒,Epstein-Barr病毒,单纯疱疹病毒,HHV-6,人类T细胞白血病病毒Ⅰ和Ⅱ型和细小病毒与LCH发病相关的证据,迄今尚无上述病毒与LCH的发病有关而令人信服的文献记载。
(二)发病机制
在过去的20年中,虽然对本病做了不少免疫学方面的检测,但尚未发现本病免疫学方面恒定性缺陷,已发现LCH损害的细胞培养有几种细胞活素如白介素Ⅰ,肿瘤坏死因子α,粒细胞-巨噬细胞无性繁殖刺激因子,白介素-8和白血病抑制因子数量显著增高,这些因子在引起(促进)LCH细胞生长上可能起着决定性的作用,但尚不明确这些细胞活素对本病不同的表现的相关性,虽然通过X-染色体灭活测定提示LCH细胞可能为无性繁殖起源,但这些细胞并不表现无性繁殖T细胞受体基因重组,这些发现结合对损害的流式细胞计数DNA分析,以及LCH可自然消退,均提示LCH为一非新生物性疾病。
鉴别诊断
1.以Letterer-Siwe 病为代表的急性播散性朗格汉斯组织细胞增生症应与脂溢性皮炎,毛囊角化病,泛发性念珠菌病等鉴别,LS病在婴幼儿期发病,全身症状及系统症状明显,皮损往往伴发紫癜,瘀点为其特点,组织病理检查可以确诊。
2.以Hand-Schüller-Christian 病为代表的慢性播散性朗格汉斯组织细胞增生症和以嗜酸粒细胞肉芽肿为代表的慢性局限性朗格汉斯组织细胞增生症的斑块性结节和黄瘤性损害应与幼年性黄色肉芽肿,播散性黄色瘤,皮肤黄色瘤,肥大细胞增生症,良性头部组织细胞增生症等鉴别。
HSC病和嗜酸性粒细胞肉芽肿免疫组化检查S-100蛋白和CDIa阳性,超微检查可见Birbeck颗粒,可与以上疾病鉴别。
朗汉斯细胞的组织细胞增多症西医治疗
治疗应根据患者年龄、疾病的范围和部位而定。
1、单系统受累者(皮肤或骨受累者)
(1)儿童仅皮肤受累,皮损持久不消退者,可局部应用氮芥水溶液(20mg/dl)共用5天。病情顽固者,可用糖皮质激素或抗有丝分裂药。
(2)成人仅皮肤受累者,局部氮芥治疗有效。亦可应用PUVA治疗。眶周嗜酸性粒细胞肉芽肿,可用CO2激光治疗。沙利度胺(反应停)亦可缓解皮损,开始100mg/d治疗1月,然后50mg/d连服1~2月。
亦有报告口服维A酸(异维甲酸)1.5mg/(kg·d)治疗1例患者使皮损完全缓解。仅有骨受累的患者,如有可能可手术切除或刮除。儿童患者可用糖皮质类固醇损害内注射,可避免外科手术损伤发生骨刺和损伤长骨生长板。尚可对成人中累及椎骨、蝶鞍和有骨折危险的持重性长骨进行放射疗法。儿童则在其他治疗失效后再考虑应用,以免长期放射治疗的副作用。
2、多系统受累者
当前最适当的治疗为硫酸长春碱(长春花碱)或依托泊苷(鬼臼乙义苷)单独或与糖皮质激素合用。硫酸长春碱(长春花碱)0.1~0.2mg/kg静脉注射,每周1次,共治疗1~3月。依托泊苷(鬼臼乙义苷)200mg/?口服或静脉注射,每3~4周中连续注射3天,至少用3~4个疗程。复发病例,用以上药物再治疗,60%患者的损害仍能完全消退。此外亦可用甲泼尼龙30mg/kg,静脉注射,连续3天作为早期治疗。对上述单一化学疗法无效者,可用硫酸长春碱(长春花碱),环磷酰胺,多柔比星(阿霉素)和苯丁酸氮芥联合治疗。对顽固性和进行性病例可用环孢素和干扰素α2治疗。同种异体骨髓移植曾成功地应用于预后很差的1位儿童。
预后
1、急性播散性朗格汉斯组织细胞增生症,如发生自发性血小板减少症和严重贫血则常致死亡。患者常有发热,病情发展迅速,总的来说预后不良。但偶亦见仅有皮损而无系统症状者,则预后良好。
2、Hand-schüller-christian病(HSCD),病程常长达数年,不经治疗约30%患者可致死亡。
3、嗜酸粒细胞肉芽肿(eosinophilic granuloma),呈慢性经过,有自愈倾向,在罕见情况下,嗜酸性粒细胞肉芽肿可发展为Hand-Schüller-christian病。
朗汉斯细胞的组织细胞增多症中医治疗
当前疾病暂无相关疗法。 以上提供资料及其内容仅供参考,详细需要咨询医生。
根据不同的症状有不同情况的饮食要求,具体询问医生,针对具体的病症制定不同的饮食标准。
本病暂无有效预防措施,早发现早诊断是本病防治的关键。
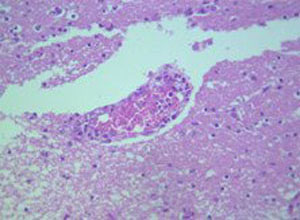
组织病理:Langerhans细胞比淋巴细胞大约4~5倍,胞核不规则,呈空泡状,核常呈肾形,有丰富淡嗜酸性胞浆,可有3种组织反应,即增生性,肉芽肿性和黄瘤性,3型组织反应之间无截然界限,可互相重叠,但组织反应类型和临床疾病分型间的关系仍较明确,一般而言,几乎纯粹的朗格汉斯细胞浸润的增生性反应是Letterer-Siwe病的典型组织表现,肉芽肿性反应是嗜酸粒细胞肉芽肿的典型表现,黄瘤性反应是Hand-Schüller-christian病的典型表现,黄瘤性一般只发生在少数器官中,因此在该病的皮损中不一定有含脂细胞。
1.皮肤增生性反应组织病理学特点
为出现广泛的朗格汉斯细胞浸润,浸润通常紧靠表皮并侵入表皮,甚至破坏表皮引起溃疡,浸润细胞大而圆,有丰富的淡嗜酸性胞浆,核常偏向一侧,核边缘有切迹或呈肾形,在一些病程急剧而致死的患者,特别是婴儿患者中,核呈多形性甚至非典型性,表现胞核大而形状不规则,染色深,在浸润细胞聚集区常见红细胞外渗,偶尔有些浸润细胞呈泡沫状,在持久存在的损害中,可见少数多核巨细胞,提示向肉芽肿过渡。
2.肉芽肿性反应
呈广泛性朗格汉斯细胞浸润,常扩展至真皮深层,有数量不等的嗜酸性粒细胞,浸润细胞常集簇成群,常见多核巨细胞,亦可见到一些中性粒细胞,淋巴样细胞和浆细胞,常见红细胞外渗,但真正的泡沫细胞不常见。
3.黄瘤性反应
表现为真皮内有大量的泡沫细胞,数量不等的朗格汉斯细胞和嗜酸性粒细胞浸润,常见多核巨细胞,主要为异物型,偶亦有呈Touton巨细胞者(含脂质的巨细胞)。
免疫组织化学检查:LCH细胞呈正常朗格汉斯组织细胞免疫表型,这些细胞表达高水平组织相容性复合物(MHC)组Ⅱ分子,S-100蛋白,CDIa复合物和CD4分子;但与Langerhans细胞不同者,LCH细胞也表达数种巨噬细胞相关标记,包括CD11C,CDW32和CD68,尚很少有可靠的标记能区分LCH细胞与正常皮肤的Langerhans细胞,但有3种标记即胎盘碱性磷酸酶(PLAP),花生凝集素(PNA)和干扰素-γ受体(IFN-γR)用来区分正常Langhans细胞与LCH细胞特别有价值。
超微检查:约50%LCH组织细胞含Birbeck颗粒。
本病呈慢性经过,有自愈倾向,在罕见情况下,嗜酸性粒细胞肉芽肿可发展为Hand-Schüller-christian病。