本病又称Dego病,Kohlmeier-Degos综合征,致死性皮肤和胃肠道细动脉血栓形成(lethal cutaneous and gastrointestinal arteriolar thrombosis)。本病是皮肤-肠道或其他器官的细小动脉内膜炎而后血栓形成的疾病,病因还不明,有认为与常染色体显性遗传,自身免疫异常和纤溶活性降低及慢病毒感染等有关。约1/3病例只有皮肤损害。约20%病例累及中枢神经系统,少见的尚可累及眼、心、肾和膀胱等,IgA可显著升高。
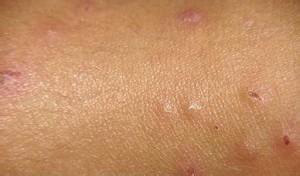
以青壮年男性发病率最高,通常是累及皮肤和肠道,而皮肤往往在先,约1/3病例只有皮肤损害,约20%病例累及中枢神经系统,少见的尚可累及眼,心,肾和膀胱等,皮肤损害主要分布于躯干和四肢,特别是在背部和肢体近端,而面和手足较少,原发损害为直径2~15mm的半球状水肿性红色丘疹,病程中部分损害持续存在或吸收消失,遗留小的白色皮肤萎缩,而大多数中央迅速坏死,发生溃疡,遗留瓷白的皮肤萎缩斑,上附有灰白色鳞屑,有狭窄的高起边缘,其上或有扩张的毛细血管,损害成批出现,少则几个,多则百余个,散在分布,较少相互融合,一般无自觉症状,可持续数个月或数年,在皮肤损害出现后3周~10余年间发生肠损害,多数是小肠,大肠,肠系膜,而胃亦有累及,呈隐袭发病,早期可仅为消化不良,腹泻或便秘等,进而发生腹绞痛和便血,最后因发展为多发性肠穿孔和腹膜炎,故预后不良,剖腹探查,可在肠壁上发现多发性卵圆形针尖至直径为2cm的浆膜下白色萎缩性瘢痕,无肠损害的患者中,有33%~50%并不产生严重后果,眼部损害为在眼睑,球结膜可有白色无血管斑,脉络膜和视网膜缺血变性,复视,视盘水肿和视神经萎缩等。
本病病因不明,有认为与常染色体显性遗传,自身免疫异常和纤溶活性降低及慢病毒感染等有关。
发病机制:发病机制还不清楚,有学者认为与常染色体显性遗传,自身免疫异常和纤溶活性降低及慢病毒感染等有关。
组织病理:主要病变为真皮下部和皮下脂肪组织内的细小动脉内膜炎,内皮细胞肿胀和增生,由于PAS阳性物质沉积而管壁增厚,血栓形成,以致产生缺血性梗死,发生楔形溃疡,中膜和外膜一般不受累,内弹力膜正常,缺血区内胶原纤维肿胀,变性或渐进性坏死,最后完全纤维化而萎缩,早期病灶内可有黏蛋白沉积,至晚期则只见于周围组织内,可见小肠浆膜下散在分布的梗死性溃疡,白色瘢痕或穿孔,其组织病理变化与皮肤损害相同。
根据临床表现和组织病理检查可以确诊。
有时需与淋巴瘤样丘疹病,急性痘疮样苔藓样糠疹和变应性皮肤血管炎相鉴别。
恶性萎缩性丘疹病西医治疗
主要是对症治疗。一般可用吲哚美辛(消炎痛)、阿司匹林与双嘧达莫(潘生丁)。虽可用皮质激素,但在晚期应注意发生肠穿孔的可能。亦可试用肝素治疗。
预后
在皮肤损害出现后3周~10余年间发生肠损害,多数是小肠、大肠、肠系膜,而胃亦有累及。呈隐袭发病,早期可仅为消化不良、腹泻或便秘等,进而发生腹绞痛和便血,最后因发展为多发性肠穿孔和腹膜炎,故预后不良。
恶性萎缩性丘疹病中医治疗
当前疾病暂无相关疗法。
(以上提供资料及其内容仅供参考,详细需要咨询医生。)
根据不同的症状,有不同情况的饮食要求,具体询问医生,针对具体的病症制定不同的饮食标准。
加强护理和营养,以提高患者的抵抗力和免疫力,预防感染应注意隔离,尽量减少与病原体的接触,避免近亲结婚,进行婚前检查,生育咨询。
免疫球蛋白尤以IgA显著升高,纤维蛋白原含量升高和血小板凝集试验为阳性,此外,一般无阳性发现。
组织活检,主要病变为真皮下部和皮下脂肪组织内的细小动脉内膜炎。
肠损害后可发生腹绞痛和便血,最后因发展为多发性肠穿孔和腹膜炎,眼部损害球结膜可有白色无血管斑,脉络膜和视网膜缺血变性,复视,视盘水肿和视神经萎缩等。