CM1神经节苷脂贮积症(CM1 gangliosidosis)是由β-半乳糖苷酶缺乏而引起的一种遗传性溶酶体疾病,其遗传方式为常染色体隐性遗传,临床特征为进行性中枢神经系统障碍及类似黏多糖贮积症Ⅰ型的骨骼异常。
-
挂什么科:内科 风湿免疫科
-
需做检查:尿常规 脑电图检查 尿液分析 脊柱椎体平扫
-
治疗方法:药物治疗 支持性治疗
-
常用药物:暂无相关信息
-
一般费用:根据不同医院,收费标准不一致,市三甲医院约(3000——8000元)
-
传染性:无传染性
-
治愈周期:15-30天
-
治愈率:60%
-
患病比例:0.00035%
-
好发人群:无特定的人群
-
相关症状:
-
相关疾病:
1.Ⅰ型又称全身性神经节苷脂贮积症,亦称婴儿型,其特点是:
(1)严重的脑变性,多于2岁内死亡。
(2)神经元,肝,脾和其他组织细胞及肾小球上皮细胞中神经节糖苷贮积。
(3)表现有Hurler病的骨骼畸形。
本病起病早,通常在出生时就可出现精神和运动障碍症状,患儿外貌严重异常,呈Hurler综合征样粗笨面容,前额突出,鼻梁扁平,眼间距增宽,齿龈肥厚,伸舌,短颈和多毛症,多数病例无角膜浑浊,但所有病人均有腰椎改变和眼底黄斑部有樱桃红斑点,还有肝脾肿大及多发性骨发育不良畸形,皮肤增厚,有时可出现毛细血管扩张,新生儿期呈蛙形体态,智力发育低下,至6~7个月时,反应迟钝,不能随物注视,甚至对外界无任何反应,肌张力减低,自主运动少,腱反射活跃,抽风发作是其突出症状,而且出现较早,用解痉药物治疗无效,抽风,反复呼吸道感染和阵发性心动过速,是病儿于2岁前死亡的主要原因。
2.Ⅱ型亦称少年型,起病较晚,新生儿期大致正常,在1岁时即出现精神,运动障碍和神经系统症状,如共济失调,步态不稳,运动不协调,抽风,语言障碍,表情淡漠,眼神呆滞及腱反射亢进等,并可随着病情的发展而逐渐恶化,Ⅱ型除起病较晚外,尚有病程较长,存活年龄较大,临床上无骨骼异常,无Hurler综合征样面容,无肝脾肿大,眼底黄斑部无樱桃红斑点等特征,视力和听觉通常不受损害,多在3~5岁夭折,常死于反复抽风及呼吸道感染。
3.Ⅲ型亦称成人型,Reuser等将Ⅳ型也归类于成人型,本型发病较晚,年幼时部分患者可有较轻的全身性神经节苷脂贮积症症状,20岁后出现进行性智力低下,口齿不清,小脑功能失调及视力减退,轻微的脊柱改变,无肝脾肿大和眼底黄斑部的樱桃红斑点,部分患者可有弥漫性血管角质瘤,全身肌张力减低,当病人躺下来或坐着松弛时,肌张力异常姿势和肌张力异常运动并不消失,磁共振显示豆状核有双侧对称性的高密度病变。
(一)发病原因
遗传基因缺陷,病人体内3种酸性β-半乳糖酶同工酶A,B和C在身体各组织中明显缺乏是本病的主要病因。
(二)发病机制
组织化学和生物化学研究证明,CM1神经节苷脂贮积症是由3种酸性β-半乳糖酶同工酶A,B和C在身体各组织中明显缺乏而引起,可分为Ⅰ,Ⅱ,Ⅲ,Ⅳ型,其发病机制和病理变化以Ⅰ型和Ⅱ型为主,Ⅰ型患儿β-半乳糖苷酶显著缺乏,该酶活性降低时,CM1神经节苷脂即沉积于体内各器官中,特别是脑灰质内,此外,该酶还参与某些酸性黏多糖的降解,故也可有部分降解的酸性黏多糖沉积于组织细胞内,其中以肝细胞内最明显,体内沉积的神经节苷脂可能来自各种细胞的细胞膜,因为该种结构中有大量神经节苷脂存在,Ⅱ型CM1神经节苷脂贮积症虽然也是β-半乳糖苷酶缺乏,但其pH活性曲线和同工酶类型与Ⅰ型有所不同。
病理:在显微镜下,Ⅰ型的病理改变是所有器官的组织细胞都有气球样空泡,以网状内皮系统细胞和神经元内最明显,细胞质包涵体组织化学染色显示溶酶体内有糖脂沉积,神经元内沉积的物质为强嗜苏丹性及PAS弱阳性,而胶质细胞和内脏细胞的组织化学染色主要显示为多糖的特征,在肝实质细胞,库普弗细胞,肾小球细胞,肾小管上皮细胞,组织细胞,造血组织的网织红细胞,心肌细胞,上皮细胞,肺和肠道的结缔组织细胞内,都可见有大的溶酶体,用电子显微镜检查,有膜的空泡内含有细小的无定形,轻度亲水性物质,偶尔含有成层的膜性结构。
在脑,小脑,脑干,脊髓及自主神经节细胞内,神经元在显微镜下呈气球样,这是由于大量具有膜性结构的溶酶体所造成的,此外,脑组织内尚有胶质反应和脱髓鞘性改变,同样,在周围血液中的单核细胞,培养的皮肤成纤维细胞,在显微镜下可有细胞质包涵体,该种包涵体,甲苯丁蓝染色呈异染色,在电子显微镜下,这两种细胞显示溶酶体增大,其内含有透明的颗粒物质。
生化分析神经元包涵体内沉积的物质,主要由CM1神经节苷脂及单涎酸衍生物,胆固醇,少量磷脂和葡萄糖神经鞘磷脂所组成,内脏器官包涵体沉积物主要由糖蛋白和单涎酸黏多糖及少量CM1神经节苷脂所组成。
Ⅱ型的病理改变与Ⅰ型相似,但程度较轻,内脏器官可以无CM1神经节苷脂沉积,而有黏多糖沉积,尿中也可排出过多的黏多糖,脑内神经元沉积物CM1脑苷脂也较Ⅰ型低。
根据临床症状,病理特点和实验室检查即可获得诊断。
Ⅰ型需与某些黏多糖贮积症鉴别,但后者病程较长,Ⅱ和Ⅲ型应与CM2神经节苷脂贮积症的婴儿型相鉴别,后者内脏不受累,必要时可通过酶的测定来进一步确诊。
CM1神经节苷脂贮积症西医治疗
治疗:
无特效疗法,主要以对症治疗为主。
预后:Ⅰ型预后较差,严重的脑变性,多数在2岁以前死亡。Ⅱ型预后较差,多在3~5岁夭折,常死于反复抽风及呼吸道感染。Ⅲ型预后较前者都好,可活到40~50岁。
CM1神经节苷脂贮积症中医治疗
暂无可参考数据。
CM1神经节苷脂贮积症日常保健
需要在医生指导下进行特殊的饮食,半乳糖来源于饮食中的奶制品,需要禁食。
CM1神经节苷脂贮积症日常预防
开展婚姻和生育指导,努力降低人群中遗传病发生率,提高人口素质。
Ⅰ型病人周围血液中淋巴细胞,单核细胞,培养的皮肤成纤维细胞,均可见有细胞内空泡,但Ⅱ型病人的阳性率低,Ⅰ型病人尿中可能有过多的糖蛋白,Ⅱ型病人尿中可排出过多的酸性黏多糖,皮肤成纤维细胞和肝组织生化分析,可见CMl神经节苷脂及硫酸角质素沉积。
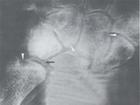
Ⅰ型最早的X线表现为长管状骨,肋骨骨膜新骨形成,继之出现椎体发育不良,并呈鸟嘴状,胸腰部脊柱侧弯,肋骨在椎体端变细,其余部分呈船桨状,还可有“乙”状形蝶鞍,髂骨翼向外张开,掌骨畸形,爪状手,关节僵直,肘关节和膝关节屈曲挛缩等,骨骼畸形在新生儿期不明显,到5~6个月时才较显著,Ⅱ型病人大多数无骨骼异常,即使出现骨骼异常症状也较轻。
Ⅰ型病人可并发腰椎改变,眼底黄斑部有樱桃红斑点,还有肝脾肿大及多发性骨发育不良畸形,皮肤增厚,有时可出现毛细血管扩张,Ⅱ型常死于反复抽风及呼吸道感染,Ⅲ型部分患者可并发弥漫性血管角质瘤,全身肌张力减低。