先天性睾丸发育不全又称Klinefetter综合征(Klinefetter syndrome),是一种发病率较高的性染色体疾病,在染色体鉴定之前,1942年Klinefelter首先报道了此症,1956年Bradbury等表明患者体细胞内呈女性X染色质,1959年Jacobs和Strong首先发现该病患者的染色体核型为47,XXY。由于性染色体异常导致睾丸发育不全,不育、智能低下等。
-
挂什么科:儿科 儿科综合
-
需做检查:染色体 羊水细胞培养染色体检查 精液检查 睾酮 胸部平片 心电图 染色体核型分析 血液生化六项检查 睾丸活检
-
治疗方法:药物治疗 手术治疗 支持性治疗
-
常用药物:暂无相关信息
-
一般费用:根据不同医院,收费标准不一致,市三甲医院约(10000——30000元)
-
传染性:无传染性
-
治愈周期:3-12个月
-
治愈率:40%
-
患病比例:0.0004%
-
好发人群:儿童
-
相关症状:
-
相关疾病:
本病的发病率相当高,但许多患者除不育外无任何症状或不适,因而不会就诊,难以发现,患者表型男性,体格较瘦长,身材较高,指间距大于身高,乳房往往增大,乳房女性化约占40%,在47,XYY及48,XXYY核型,具有2个Y染色体患者高体型表现更为明显,青春期发育常延缓,由于无精子,一般不能生育(偶有例外)。
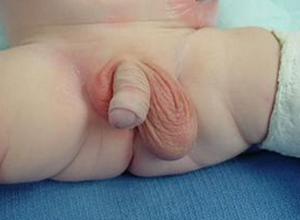
体格检查发现男性第二性征不明显,无胡须,无喉结,皮肤白皙,睾丸小,阴茎亦小,可有隐睾或尿道下裂,阴毛发育差。
患者可有性格孤僻,腼腆,不活跃,胆小,缺乏男孩性格,在标准47,XXY核型中,约有25%显示中等度智能发育落后,表现为语言和学习障碍,此征在青春期前缺乏明显症状,不易认识,若对智能落后或行为异常的儿童做常规的染色体核型分析,可进行早期诊断。
先天性睾丸发育不全在儿童期可因隐睾或者小睾丸来诊,但多数因症状不严重,缺乏特征性或体格检查疏忽不易被重视,一般在青春发育期,由于睾丸不发育,男性化不全,部分患者有女性乳腺发育或不育而来诊。
(一)发病原因
本病征系性染色体异常,多数患者多了一个X染色体,故最常见的外周血白细胞染色体核型是47,XXY,或为嵌合体47,XXY/46,XX或47,XXY/48,XXXY,甚至有更多的X染色体,如49,XXXXY,口腔黏膜X小体检查阳性者在XXY核型中占93%。
本病征的各种核型是由于细胞成熟分裂或受精卵在卵裂中发生的性染色体或性染色单体不分离的结果,从染色体基因标记研究示卵子细胞不分离多于精子的2倍,上述性染色体畸形,以高龄妇女妊娠中机会为多,可因卵细胞的衰老,着丝点纵裂动力减弱或纺锤丝迷向的缘故,有迹象示本病征中X染色体数目愈多睾丸曲细精管玻璃样变性,间质增生纤维化愈严重,智力发育亦愈受累,其机制不明,曾有学者推论X染色体的基因对睾丸发育有不利影响,因此本病征主要病理特点为曲精细管发育不全,小睾丸,不能产生精子或精子极稀少,故不生育,间质细胞呈腺瘤样丛集,其细胞内所含脂类物质及分泌颗粒减少,有些病例有脑电图检查异常,提示有轻微器质性脑功能损害,此系原发抑或继发于内分泌功能紊乱尚无定论。
(二)发病机制
先天性睾丸发育不全是一种先天性睾丸生精发育不全或不发育的疾病,患者常因不育或体检外生殖器不发育而就诊,然后经染色体检查确诊。
该病不同类型的共同特征是性染色体比正常的XY多1个或1个以上的X染色体,多余的X染色体对睾丸及体征均有不良影响,尤其对体征影响更甚,X染色体越多,睾丸发育不良程度越明显,症状越严重,智能发育越差,其他畸形也往往愈多,由于Y染色体有睾丸决定基因(TDF),该病患者均有Y染色体,因此患者表型为男性,但超过正常的X染色体导致不同程度的女性化。
先天性睾丸发育不全的染色体核型较多,1987年在国内王德芬等报道的62例中,47,XXY占71.0%,47,XXY/46,XY嵌合体占24.2%,48,XXXY及48,XXYY各约3.2%和1.6%,这些核型的形成是由于细胞成熟分裂,或受精卵在卵裂中发生的性染色体或性染色单体不分离的结果,染色体基因标记研究提示,卵子细胞的不分离多于精子的2倍,性染色体畸变,以高龄孕妇妊娠中机会为多,可因卵细胞的衰老,着丝点纵裂动力减弱或纺锤丝迷向的缘故,导致亲代在生殖细胞形成过程中发生了性染色体不分离,有分析表明,60%的患者是由于母体染色体不分离,40%是由于父体染色体不分离所致,成熟分裂过程中染色体不分离约有83%的可能发生在第1次减数分裂,17%的可能发生在第2次减数分裂。
由于X染色体的增多,致使睾丸未发育,阴茎短小,血浆睾酮降低,FSH,LH升高;雄性激素分泌不足,FSH升高可能是因为支持细胞损伤,分泌抑制素减少之故,睾酮水平偏低,说明该病患者睾丸间质细胞分泌睾酮功能降低,这就必然使LH代偿性升高,通过电镜观察,患者睾丸的间质细胞内有异常线粒体和内质网,这可能是干扰睾酮生物合成障碍的物质基础。
诊断
根据临床特征怀疑为先天性睾丸发育不全的患者可先予生化检查,若血清FSH,LH增高,睾酮水平较正常为低,可对患者作外周血淋巴细胞染色体核型分析,确定X染色体数目,是诊断先天性睾丸发育不全的主要依据,FISH技术是快速诊断方法。
鉴别诊断
本征要与青春期发育延迟做鉴别诊断,先天性睾丸发育不全在青春期血促性腺激素明显升高,睾酮水平较低,而青春期发育延迟者处于未发育水平,无促性腺激素升高,如对本征患者在青春期后作睾丸活体组织检查,可见曲细精管玻璃样变,其睾丸间质细胞(Legdig细胞)虽有增加,但内分泌活力不足。
小儿先天性睾丸发育不全西医治疗
时机很重要,成年人的治疗效果比较低。及早发现,及早治疗。
患者自11~12岁开始应进行雄激素疗法。一般应用环戊丙酸酯(cyclopentylproplonate ester),开始每次肌注50mg,每3周1次,每隔6~9个月增加剂量50mg,直至达到成人剂量(每3周250mg)。对较年长的病人,开始剂量和递增用量均可加大以便收效较快。国内目前较常用的是十一烷酸睾酮(testosterone undecanoate)又称安雄(andriol),为睾酮的衍生物,有较强的雄激素作用和蛋白同化作用。油剂针剂每支250mg,每月注射1次,连续4个月。注射后通过淋巴系统缓慢吸收,血药浓度峰值时间为2天,药效可维持1个月。口服剂型每粒40mg,易被胃肠道吸收,2.5~5h达高峰,10h后恢复原先水平。口服起始量为120~160mg/d,分3次服用,连续2~3周,然后改为维持量40~120mg/d。由于十一酸睾酮通过淋巴系统吸收,不经过肝脏,故对肝功能无影响。
如果内分泌治疗无效,可靠手术治疗。手术治疗后应随访,每年一次,直至青春期。
对于不育者,伴有精液常规异常的精索静脉曲张者需行精索静脉高位结扎术,隐睾或睾丸下降不全者可行睾丸下降固定术,以促进睾丸的生精功能。保持输精管道的通畅 积极治疗性功能障碍,保持正常的性生活。对于先天性输精管 缺如、输精管道梗阻和输精管结扎者应积极手术治疗。
预后
自青少年期起辅以雄激素治疗,常可见其学习技能水平提高,与正常儿差距缩小,变得开朗,自信力增强,但停止治疗后有的又复后退。
先天性睾丸发育不全患者根据其染色体核型可初步判断其预后,核型中X染色体越多,预后越差,其次早期诊断,尽早治疗对患儿症状的改善也很有关系。
小儿先天性睾丸发育不全中医治疗
中医认为肾主藏精,肾主生殖、发育等等,用填补肾精的办法,鼓舞肾气,对于治疗睾丸发育不全有一定的治疗意义。
以上提供资料及其内容仅供参考,详细需要咨询医生。
日常保健
注意食品的选择,应供给营养丰富、清淡易消化的食物,如藕粉、牛奶、蛋羹、米粥等。平常也要保证营养及进食量,少吃油腻或过甜的食物,避免吃生冷食品。如对某些食物过敏就应忌口,.禁止近亲结婚,避免高龄妊娠。.婚前检查以期发现不应结婚的遗传病或其他疾病。
先天性睾丸发育不全的染色体畸形,以高龄妇女妊娠中机会为多。预防措施包括避免近亲结婚、携带者基因检测、产前诊断和选择性人工流产等防止患儿出生。
1、禁止近亲结婚,避免高龄妊娠。
2、婚前检查以期发现不应结婚的遗传病或其他疾病。婚前体检在预防出生缺陷中起到积极的作用,作用大小取决于检查项目和内容,主要包括血清学检查(如乙肝病毒、梅毒螺旋体、艾滋病病毒)、生殖系统检查(如筛查宫颈炎症)、普通体检(如血压、心电图)以及询问疾病家族史、个人既往病史等,做好遗传病咨询工作。在妊娠期产前保健的过程中需要进行系统的出生缺陷筛查,包括定期的超声检查、血清学筛查等,必要时还要进行染色体检查。
3、携带者的检出 通过群体普查、家系调查及系谱分析、实验室检查等手段确定是否为遗传病,并确定遗传方式等。
4、遗传咨询
对于有家族病史的:要遗传咨询可以帮助患病成人进行选择性生育。由于大多数病例是由未患病父母发生了完全不能预测的基因突变所引起的。所以在妊娠期产前保健的过程中需要进行系统的检查。
5、产前诊断 产前诊断或宫内诊断,是预防性优生学的一项重要措施。所用产前诊断技术有:①羊水细胞培养及有关生化检查(羊膜穿刺时间以妊娠16~20周为宜);②孕妇血及羊水甲胎蛋白测定;③超声波显像(妊娠4个月左右即可应用);④X线检查(妊娠5个月后),对诊断胎儿骨骼畸形有利;⑤绒毛细胞的性染色质测定(受孕40~70天时),预测胎儿性别,以帮助对X连锁遗传病的诊断;⑥应用基因连锁分析;⑦胎儿镜检查。
6、羊水细胞染色体核型分析:为了防止先天性性染色体疾病患儿的出生,在妊娠中期第16~20周行羊膜腔穿刺,抽取羊水细胞,经培养后进行胎儿染色体核型分析,发现异常核型及时终止妊娠,可降低出生缺陷。
7、造成先天性疾病的原因甚为复杂,包括妊娠期间的感染、高龄生育、近亲婚配、辐射、化学物质、自体免疫、遗传物质异常等。
早诊断早治疗是本病的防治关键。
1.细胞遗传学检测
(1)口腔黏膜X染色质检查:X染色质也称X小体(Barr体),正常情况下X小体的数目等于X染色体数目减去1,正常男性由于仅有1条X染色体,故X小体检查呈阴性,而该病患者有2条或2条以上X染色体,故他们的口腔黏膜细胞中可检查到1个或1个以上X小体,该检查方法简单,涂片后镜检即有结果,可作为性染色体异常的初筛试验,无细胞培养条件的地方也可进行,但不能做正确的核型分析。
(2)染色体核型分析:
①外周血淋巴细胞染色体核型分析:正常男性体细胞中的性染色体为XY,该病性染色体标准型为XXY,为性染色体三体型,该病80%核型为标准型47,XXY或者标准型核型变型,例如48,XXXY。48,XXYY。49,XXXXY。49,XXXYY,15%为嵌合体型,嵌合体有47,XXY。46,XY。47,XXY。46,XX。47,XXY。46,XY。45,X。47,XXY。46,XY。46,XX等。
②羊水细胞染色体核型分析:为了防止先天性性染色体疾病患儿的出生,在妊娠中期第16~20周行羊膜腔穿刺,抽取羊水细胞,经培养后进行胎儿染色体核型分析,发现异常核型及时终止妊娠,可降低出生缺陷。
2.荧光原位杂交
妊娠中期对胎儿染色体核型分析,无论是外周血淋巴细胞还是羊水细胞均需作细胞培养后才能进行核型分析,因此很费时间,而荧光原位杂交检测不需细胞培养,可直接对间期细胞进行杂交检测,缩短了诊断时间,用于产前诊断则该方法更显出其优势,可直接和绒毛细胞,羊水细胞杂交,1天即可得出结论,若有异常,即可终止妊娠,提早结束妊娠的时间。
3.生化检验及其他检验
患者血清中睾酮降低,对下丘脑垂体反馈抑制减弱,垂体促性腺激素黄体生成激素(LH),尿促性素(FSH)升高,血清睾酮水平较正常为低;促性腺激素释放激素(LHRH)兴奋试验显示FSH反应增高,LH反应正常;人绒毛膜促性腺激素(HCG)刺激试验睾酮呈低反应。
患者精液中无精子生成,睾丸活体组织病理检查见曲细精管变性,间质细胞增生。
腹部B超检查,心电图和X线胸片检查,了解有无其他畸形和异常。
1、雄激素缺乏:类无睾症体型,身材正常或偏高,下肢较长,阴茎正常或短小、性功能低下,约97%的患者为不育症,骨质疏松和肌肉力量降低。
2、高达13%的患者合并静脉曲张、血栓、慢性腿部溃疡,可能是由于雄激素缺乏导致的纤维蛋白溶解降低,而非血管解剖学改变。
3、学习、语言、智力方面障碍。