本病为神经皮肤综合征(neurocutaneous syndrome)中最常见的一种综合征,是一些起源于外胚层的组织和器官发育异常的疾病。常表现为神经、皮肤和眼睛的异常,有时也波及中胚层或内胚层发育的器官。由于受累的器官、系统不同,临床表现多种多样。目前已知此类疾病多达40余种,但常见的只有3种,即神经纤维瘤病,结节性硬化症及脑面部血管瘤病(Sturge-Weber综合征), 这些疾病多表现为常染色体显性遗传,有一个较高的、不完全的外显率。目前对这类疾病的病因了解得还不清楚,可能与胚胎发育早期出现某些变异有关。
-
挂什么科:儿科 小儿外科
-
需做检查:核磁共振成像(MRI) 电测听 脑电图检查 脑诱发电位 颅脑CT检查 裂隙灯 头颅平片
-
治疗方法:对症治疗 药物治疗 支持治疗
-
常用药物:暂无相关信息
-
一般费用:根据不同医院,收费标准不一致,市三甲医院约(5000——10000元)
-
传染性:无传染性
-
治愈周期:2-4周
-
治愈率:80%
-
患病比例:0.003%
-
好发人群:儿童
-
相关症状:
-
相关疾病:
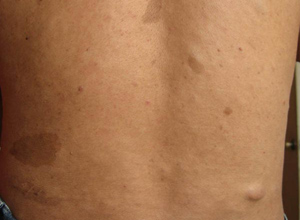
1.皮肤改变: 咖啡牛奶斑是本病的重要体征,出生时即可发现,为一些浅棕色(咖啡里加牛奶的颜色)斑,大小不等,形状不一,不隆起于皮肤,不脱屑,感觉无异常,除手掌,足底和头皮外,躯体其他部位均可波及,正常小儿有时也可见到1~2块咖啡牛奶斑,无诊断意义,6块以上直径大于5mm的咖啡牛奶斑则有诊断价值,有时在腋窝鼠蹊部或躯干其他部位见到一些1~3mm直径似雀斑状的浅棕色斑,称为腋窝雀斑,成簇出现,数目往往较多,也具有诊断意义。
皮肤的神经纤维瘤在婴幼儿时期往往不明显,青春期后增多,表现为结节状隆起,有时有蒂,与皮肤色泽一致或呈暗红色,大小由数毫米至数厘米,数目多少不等,多见于躯干,四肢及头部较少,如肿瘤压迫神经可引起疼痛或功能障碍, 丛状神经纤维瘤常波及面部,儿童期也可见到,常破坏面容,颈部或纵隔的丛状神经纤维瘤可压迫呼吸道,影响呼吸。
2.眼部异常: 虹膜部位常可见到色素性虹膜错构瘤(Lisch小体),一般体格检查不能发现,需在裂隙灯下观察,表现为突起的褐色斑块,边缘清晰,无特殊症状,6岁以后常见,有诊断意义。
3.神经系统: 神经纤维瘤组织在病理上属错构瘤样结构,为良性肿瘤,身体各部位的神经纤维均可受累,由于肿瘤的性质,部位的不同临床表现也多种多样,视神经胶质瘤可见于15%的病人中,表现为进行性视力丧失,视神经萎缩,局部疼痛或眼球突出,单侧或双侧,听神经瘤往往在10岁以后发生,表现为听力丧失,耳鸣,眩晕及面肌无力,脑部还可见到脑膜瘤,星形细胞瘤及室管膜瘤,脊髓及神经根也可波及,本病可伴有学习困难及行为障碍,但明显智力低下及癫痫发作不太多见。
4.其他系统: 骨骼常表现为先天性骨发育不良,骨皮质变薄,钙化不全等,常可见蝶骨发育不良,病理性骨折,胫骨假关节形成等。
1.Ⅰ型神经纤维瘤病 具有下列2项或2项以上:
(1)6个或6个以上咖啡牛奶斑,青春期前直径大于5mm,青春期后15mm。
(2)腋窝雀斑。
(3)视神经胶质瘤。
(4)2个以上神经纤维瘤或1个丛状神经纤维瘤。
(5)一级亲属中有Ⅰ型神经纤维瘤。
(6)2个或更多的Lisch小体。
(7)骨病变。
2.Ⅱ型神经纤维瘤病
(1)双侧听神经瘤(需经MRI,CT或组织学证实)。
(2)一侧听神经瘤,同时一级亲属中有Ⅱ型神经纤维瘤病。
(3)一级亲属中有Ⅱ型神经纤维瘤病,且患者本人有下列任何两种疾病:神经纤维瘤,脑(脊)膜瘤,神经鞘瘤,神经胶质瘤。
(一)发病原因
为常染色体显性遗传,外显率变异性较大,目前已知本病在染色体上的基因定位,Ⅰ型在17染色体上(17q11);Ⅱ型在22染色体(22q11→13),多发性神经纤维瘤病为胚胎形成早期阶段神经嵴分化和迁徙异常导致的多系统损害的,常见的常染色体显性遗传病。
(二)发病机制
本病变化多端,各个系统及器官均可受累,出生即有,呈慢性进展特征,并发症多出现于10年后,目前根据临床表现,细胞生物学和分子生物学特点将其分为Ⅰ型神经纤维瘤病(neurofibromatosis typeⅠ,NFⅠ)亦称von Recklinghausen病和Ⅱ型神经纤维瘤病(neurofibromatosis typeⅡ,NFⅡ),即双侧听神经瘤。
1.Ⅰ型神经纤维瘤病: 主要病理特征为沿着粗大的末梢神经生长的肿瘤(即分布于脊神经,脑神经,皮肤或皮下神经的多发性神经纤维瘤及神经鞘瘤),肿瘤大小不一,与神经鞘膜紧密联结,附有中胚层的神经束膜和外膜的细胞,皮肤或皮下神经纤维瘤大多数位于真皮和侵入皮下组织,病变界线不清,肿瘤无包膜,内有成熟的新生胶原纤维,无髓鞘及有髓鞘的纤维轴索掺杂,并可见到成团的施万细胞,中枢神经系统亦可见肿瘤发生,最常见的是视神经胶质瘤,在基底节和丘脑部位也可出现胶质瘤,在Ⅱ型中为听神经瘤及脑膜瘤,也可出现其他肿瘤,如Wilms瘤,成神经细胞瘤和嗜铬细胞瘤等,一般来说不论中枢神经或是末梢神经的神经纤维瘤多为良性,但也可恶性变,大约有5%转变为神经纤维肉瘤。
2.Ⅱ型神经纤维瘤病: 听神经瘤病理改变属前庭神经鞘瘤,NFⅡ常合并有脑膜瘤,脊膜瘤,星形细胞瘤以及脊旁后根神经鞘瘤,皮肤肿瘤以神经鞘瘤为主,偶有皮肤神经纤维瘤,极少出现丛状神经纤维瘤。
本症应与Watson综合征,骨纤维结构不良综合征,局部软组织蔓状血管瘤等相鉴别。
小儿神经纤维瘤病西医治疗
当肿瘤压迫神经系统有临床症状时,可行手术切除,放射治疗无效。
1.Ⅰ型神经纤维瘤病 无特殊治疗,当肿瘤生长迅速且有剧痛时应切除以防恶变,对丛状神经纤维瘤和中枢型神经纤维瘤,应尽早施行手术,蔓状神经纤维瘤病变部位多为小蔓状血管窦样改变,血管极为丰富,因此手术时应注意在病变外正常组织处切口,彻底切除肿瘤及其周围组织,术后还可利用激光技术“清扫”,以防复发,单侧眶板缺如者可予修补,癫痫发作者应予抗痫治疗。
2.Ⅱ型神经纤维瘤病 本病主要是对症治疗,手术治疗效果差,容易复发,因此原则上这类病人不适于做手术治疗,较小的肿瘤可采用聚集放射疗法,深部X射线及镭放射治疗无效,而肿瘤大到威胁病人生命时应考虑手术。
预后
目前尚无有效的治疗措施能阻止或逆转NFⅠ的病程;Ⅱ型神经纤维瘤病 症手术治疗效果差,容易复发,因此,防治关注的焦点集中在基因诊断和基因治疗。
小儿神经纤维瘤病中医治疗
当前疾病暂无相关疗法。
(以上提供资料及其内容仅供参考,详细需要咨询医生。)
注意平时的生活习惯,注意多吃一些营养含量高的食物。
通过对某一改变的DNA序列进行特征性单链构象多态性分析,为检查胎儿DNA提供了精确的产前检查方法,在家族病例中(有受累及未受累的家族成员)利用连锁分析加之产前诊断,能达到一定精确程度,必要时可终止妊娠。
目前已应用一种直接基因诊断技术-蛋白截断分析(protein truncation assay),结合基因连锁和突变分析可定出很多NF Ⅰ基因的突变株,使对NF Ⅰ的基因诊断和产前诊断成为可能,NFⅠ基因定位于染色体17q11.2,随着突变检测技术的完善,即基因扩增和等位基因特异性寡核苷酸杂交法,限制性长度片段多态性连锁分析,蛋白截断分析法,错配化学断裂法及变性梯度凝胶电泳法的综合应用可提高NFⅠ产前诊断和症状前诊断的准确率和可靠性,NFⅡ的多数患者是突变的结果。
必要时可做皮肤和皮下结节或神经干包块的病理活检。
1.基础检查: 临床考虑本病患儿应作如下基础检查,如电测听,脑干诱发电位,视觉诱发电位,脑电图,心理测验(包括预测学习能力)等,可发现脑干听觉诱发电位等各种异常。
2.X射线照片: 头颅内听道X射线片示双侧内听道破坏;骨骼X射线检查有助于发现骨骼畸形,该病骨骼X射线片上可见大的,多发的囊状结构,可见透明区,骨皮层变薄。
3.椎管造影: 有助于发现中枢神经系统肿瘤。
4.MRI成像: 头颅MRI成像示双侧听神经瘤,在苍白球,丘脑和内囊部位显示不正常的信号,这可能意味着存在低恶性度的神经胶质瘤或错构瘤,而CT扫描则检测不到,这些可以是学习困难,注意力不集中和语言障碍的原因,MRI示大脑白质斑块状异常信号,TW1低信号,TW2高信号,性质不明,暂称之为不明原因发光物质,无症状病人应每年反复查体,进行神经学评价,包括血压,听觉和视觉的筛查,探查神经纤维瘤病的并发症。
5.脑或视神经CT扫描: CT可发现中枢神经系统肿瘤,但约85%患者颅脑CT无异常,脑室大小正常的巨头很常见,继发于导水管狭窄的脑水肿则少见。
神经纤维瘤可能阻塞脑血管致使偏瘫和智力障碍,由于本病的严重性和不确定性,所以存在心理障碍是不足为怪的,NFⅠ病人中恶性肿瘤也是值得注意的问题,神经纤维瘤偶尔分化成神经纤维肉瘤或恶性神经鞘瘤;嗜铬细胞瘤,横纹肌肉瘤,白血病和Wilms病的发病率比一般人群高,然而,在NFⅠ患者中中枢神经系统肿瘤(包括视神经胶质瘤,脑和脊髓的脑脊膜瘤,神经纤维瘤,星形细胞瘤和神经鞘瘤)发生率很高,故其是患者发病和死亡的重要原因。
肿瘤压迫神经可引起功能障碍,压迫呼吸道影响呼吸,发生进行性视力丧失或眼球突出,听力丧失,耳鸣,眩晕,伴有学习困难及行为障碍,常可见病理性骨折,胫骨假关节形成等,少数可伴发惊厥,语言和运动发育迟缓。