小儿Alstrom综合征又称先天性黑蒙,遗传性先天性视网膜病、Alstrom-Olsen综合征等。属于常染色体隐性遗传性疾病,主要为视力减退、神经性耳聋、肥胖、糖尿病、尿崩症、肾功能不全、性腺功能低下、高尿酸血症及高甘油三酯血症等。是发生最早、最严重的遗传性视网膜病变, 出生时或出生后一年内双眼锥杆细胞功能完全丧失,导致婴幼儿先天性盲。该病因缺乏氨基己糖苷酶A,不能裂解神经节苷分子末端的N-乙酰半乳糖胺,导致神经节昔脂在脑组织中积蓄致病。故又名神经节苷脂累积病。小儿Alstrom综合征没有有效疗法。按糖尿病、尿崩症等常规用药均无效。预后不良,患者常于3、4岁前死于呼吸道感染。
主要为视力减退,神经性耳聋,肥胖,糖尿病,尿崩症,肾功能不全,性腺功能低下,高尿酸血症及高甘油三酯血症等,肥胖一般始于婴幼儿期,躯干型,2~10岁最显著,自幼烦渴,多饮,多尿,食欲亢进。
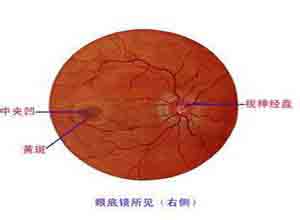
视力呈进行性减退是本病征恒定症状,常始于2岁,两眼轻度内斜,眼底示双侧原发性视神经萎缩,听力轻度减退亦为本病征恒定表现,听力测定示为中度神经性耳聋,患儿智力正常,静脉肾盂造影示双侧肾盂输尿管扩张,糖尿病也为常见的内分泌代谢紊乱,常有高尿酸血症及高三酰甘油血症,本病征无多指畸形,或精神发育迟缓,智力低下等改变,是与其他病征不同之处。
(一)发病原因
本病征病因未明,为常染色体隐性遗传性疾病。
(二)发病机制
本病呈常染色体隐性遗传,氨基己糖苷酶A基因座位位于15q23~q24,同胞再发风险率为1/4,近二三十年来病因,生化研究,诊断方法等方面都更趋完善,本病因缺乏氨基己糖苷酶A,不能裂解神经节苷分子末端的N-乙酰半乳糖胺,导致神经节昔脂在脑组织中积蓄致病,故又名神经节苷脂累积病。
根据临床症状及实验室检查应考虑本征。
以出生或出生后不久的进行性减退伴有眼球震颤等表现而区别于早发性视网膜色素变性,临床上须与Laurence-Moon-Biedl综合征,Walfran综合征,Refsum综合征及家族性肾炎等疾病相鉴别。
小儿Alstrom综合征西医治疗
1、治疗
小儿Alstrom综合征目前没有有效疗法。按糖尿病、尿崩症等常规用药均无效。有报道称自人胎盘中提取氨基己糖苷酶A制成针剂注射,尝试治疗本病,但尚未取得满意效果。
2、预后
小儿Alstrom综合征预后不良。患者常于3、4岁前死于呼吸道感染。
小儿Alstrom综合征中医治疗
暂无可参资料。
日常保健
加强妊娠期保健,避免疾病患儿的出生。
健康教育
本病征为常染色体隐性遗传性疾病,病因未明。检查血液中氨基己糖苷酶A活性,可检查出患者和携带者。对培养的羊水细胞生化分析、可做出产前诊断。
预防措施应从孕前贯穿至产前:
婚前体检在预防出生缺陷中起到积极的作用,作用大小取决于检查项目和内容,主要包括血清学检查(如乙肝病毒、梅毒螺旋体、艾滋病病毒)、生殖系统检查(如筛查宫颈炎症)、普通体检(如血压、心电图)以及询问疾病家族史、个人既往病史等,做好遗传病咨询工作。
孕妇尽可能避免危害因素,包括远离烟雾、酒精、药物、辐射、农药、噪音、挥发性有害气体、有毒有害重金属等。在妊娠期产前保健的过程中需要进行系统的出生缺陷筛查,包括定期的超声检查、血清学筛查等,必要时还要进行染色体检查。
一旦出现异常结果,需要明确是否要终止妊娠;胎儿在宫内的安危;出生后是否存在后遗症,是否可治疗,预后如何等等。采取切实可行的诊治措施。
所用产前诊断技术有:①羊水细胞培养及有关生化检查(羊膜穿刺时间以妊娠16~20周为宜);②孕妇血及羊水甲胎蛋白测定;③超声波显像(妊娠4个月左右即可应用);④X线检查(妊娠5个月后),对诊断胎儿骨骼畸形有利;⑤绒毛细胞的性染色质测定(受孕40~70天时),预测胎儿性别,以帮助对X连锁遗传病的诊断;⑥应用基因连锁分析;⑦胎儿镜检查。
通过以上技术的应用,防止患有严重遗传病和先天性畸形胎儿的出生。
尿糖酮体阴性,空腹血糖5.8~8.6mmol/L,糖耐量显著降低,尿酸464.0μmol/L(7.8mg/dl),血三酰甘油1.70mmol/L(150mg/dl),β-脂蛋白365mg/dl,胆固醇4.4mmol/L(170mg/dl),检查血液中氨基己糖苷酶A活性,可检查出患者和携带者,对培养的羊水细胞生化分析,可做出产前诊断。
眼底示双侧原发性视神经萎缩,听力测定示轻度神经性耳聋,静脉肾盂造影示双侧肾盂输尿管扩张。
糖尿病,尿崩症,肾功能不全,性腺功能低下,高尿酸血症及高三酰甘油血症等,并发呼吸道感染。