反应性穿孔性胶原病(reactive perforating collagenosis)罕见,是一种表现为胶原经表皮排除的穿通性皮肤病,由Mehregan于1967年首先报道。本病病因未明,遗传因素和创伤是两个重要的致病因素,儿童多发,最早发病年龄为9个月。成人期发病的患者往往合并有严重的糖尿病、慢性肾衰竭、肝病等,全身疹痒严重,此型又称为获得性反应性穿通性胶原病(ARPC)。
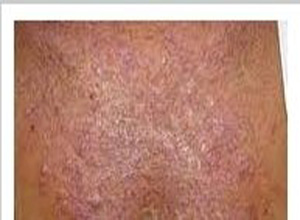
皮肤损害为帽针头大小角化性丘疹,逐渐增大,直径约5~6mm,正常肤色,表面光滑,中心脐窝状下陷,嵌有棕褐色角栓物(不易剥脱),皮疹排列呈线状群集,约4~6周能自然消退,残留暂时性色素减退,不久于其旁可出现新疹,好发于12岁以内儿童,多见于躯干,四肢及面部,轻微痒。
(一)发病原因
发病常与外伤有关,家族史明显,可能为常染色体隐性遗传。
(二)发病机制
发病机制还不确切。
根据临床表现,皮损特点,组织病理特征性即可诊断。应与皮肤穿入性毛囊和毛囊周角化病(Kyrle病),穿通性毛囊炎及匐行性穿通性弹力纤维病鉴别。
反应性穿孔性胶原病西医治疗
对于本病的治疗目前尚无特效治疗方法,如有明确的诱因需积极进行治疗。Gambichler等研究在反应性穿通性胶原病中TGF-β3和细胞外基质蛋白(如MMP-1、 TIMP-1)上调,其共同作用是参与组织创面的修复。陈小敏等报道1例用0.1%维A酸乳膏治疗有效的病例。Sehgal和Sumiyuki Mii,报道2例用NB-UVB治疗本病皮疹消失,2年内未复发。Hoque等报道了3例别嗓醇治疗本病有效的病例。目前没有统一的治疗方案,本病皮疹亦可自发消退,消退过程中丘疹变平,中心脐凹由变浅到消失,角栓自然脱落,并留有暂时性色素减退区,一般不引起皮肤萎缩。本例患者疹痒和搔抓可能是诱因,确切病因尚待研究。
反应性穿孔性胶原病中医治疗
当前疾病暂无相关疗法。
(以上提供资料及其内容仅供参考,详细需要咨询医生。)
根据不同的症状,有不同情况的饮食要求,具体询问医生,针对具体的病症制定不同的饮食标准。
本病暂无有效预防措施,早发现早诊断是本病防治的关键。
组织病理:早期表皮变薄,呈杯状凹陷,真皮乳头增宽,胶原纤维嗜碱变性,晚期杯状损害下方可见角化不全,细胞碎片及变性胶原纤维构成角栓,穿过杯底嵌入杯中,表皮坏死,不含弹力纤维,真皮上部毛细血管数量增加,少许淋巴细胞和组织细胞浸润。
在组织病理上本病需与其他类型穿通性皮肤病如葡行性穿通性弹力纤维病、穿通性毛囊炎、穿通性环状肉芽肿及Kyrle病相鉴别。葡行性穿通性弹力纤维病的组织病理表现可见狭长的穿通表皮或毛囊角栓,由角化不全性角质、嗜苏木紫碎片和弹性纤维碎片组成;穿通性毛囊炎的组织病理表现为毛囊口扩张,内填角化不全性角栓,并含有嗜苏木紫碎片和卷曲的毛发;穿通性环状肉芽肿的组织病理表现为真皮胶原纤维变性,并有渐进性坏死,坏死区周围有上皮样细胞核慢性炎细胞。Kyrle病的组织病理表现为角化不全的角栓填人凹下部分,含有嗜苏木紫碎片而无弹性纤维组织,在穿通的基底部有肉芽肿性炎症反应。
发病常与外伤有关,家族史明显,可能为常染色体隐性遗传,由于本病可伴有瘙痒,且皮肤完整性被破坏,故可因患者抓挠诱发皮肤细菌感染或者真菌感染,通常继发于体质低下,或长期使用免疫抑制剂以及有灰指甲等真菌感染的患者,如并发细菌感染可有发热、皮肤肿胀、破溃及脓性分泌物流出等表现。