马方综合征是因先天性中胚层发育不良,过度增殖,增生的中胚叶组织营养障碍所致。典型临床表现为矮胖身材,粗短指趾,宽胸,皮下脂肪丰满,肌肉发育良好,智力正常,晶状体异位,小球形晶状体。又称为Marchesani综合征、蜘蛛指征、肢体细长症,短指-晶状体脱位综合征。其特征是周围结缔组织营养不良,骨骼异常、内眼疾病和心血管异常,是一种少见的伴有全身发育异常的遗传性疾病。
生后即可发现,据统计,发病率女多于男。
1.骨的异常 身高而细,头颅长且窄,肢体长,大腿及前臂为明显,指距超过身长,尤以手指,足趾细长如蜘蛛脚样,足部常有明显外翻,个别有锤状指畸形,胸廓常呈漏斗胸伴翼状肩胛骨,有时为扁平胸,脊柱后突,侧弯,常限于胸椎。
2.韧带和关节松弛 肌张力明显减低,关节过度伸展,腕部征阳性(即病人一手握住另一手腕部,大拇指放在桡骨茎突上,大拇指与小指不用力而形成一个圈,正常人大拇指与小指间有距离,不易形成环),屡发性髌骨脱位和髋关节自然脱位也不少见,也有腹股沟疝和横膈疝。
3.外貌 面容憔悴,无力,但智力正常,耳大且位置较低,外耳多有畸形,高腭弓,眉弓较突起,显得眼下陷。
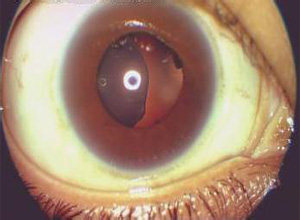
4.眼部表现 本病征50%~80%的患儿有眼部变化,常见的有晶状体脱位或半脱位,白内障,高度屈光不正,角膜大小变异和混浊;少见的有青光眼,葡萄膜色素分布异常,眼球凹陷,眼裂倾斜,弱视,色盲等。可以引起失明。
5.心血管改变 较常见,Even 1940年复习文献,1/3病例可有心血管改变,如二尖瓣反流,房间隔缺损,室间隔缺损,动脉导管未闭,法洛四联症等。
6.其他异常 有第二性征发育不良,耳壳畸形,牙齿细长等,尚可有肺分叶异常,肺叶发育不全,游走肾,输尿管狭窄等异常。
(一)发病原因
本病呈常染色体显性遗传,Dietz等(1991)通过家族的连锁分析,将本病基因定位于15q15~q21.3,在人体很多组织如心内膜,心瓣膜,大血管,骨骼等处,均有硫酸软骨素A或C等黏多糖堆积,从而影响了弹力纤维和其他结缔组织纤维的结构和功能,使相应的器官发育不良及出现功能异常,Abraham等(1982)提出,主动脉弹性蛋白有异常,桥粒蛋白和异桥粒蛋白减少,而赖氨酰残基相应增加,是该病的主要改变,病人尿中羟脯氨酸排泄量有增高,血中黏蛋白和黏多糖也增高。
(二)发病机制
通过家族的连锁基因定位显性遗传,可以从病人尿中羟脯氨酸排泄量增多来证明,本病为弹力纤维缺损,亦即胶原代谢异常,结缔组织纤维是机体组织结构中很重要的成分,因此,当它发生异常时,便会影响全身的脏器(中胚层组织),尤以骨骼和心血管系统更为显著,在蜘蛛指以及凹陷的胸部或舟状胸部都表示了四肢管状骨,手指和肋骨纵轴过度增长,可能是由于骨膜纤维成分缺陷的结果,在主动脉和肺动脉中层有酸性黏多糖沉积,本病有家族倾向,呈常染色体显性遗传。
病理:肉眼改变有眼的异常,升主动脉扩张,慢性主动脉夹层瘤形成,心脏瓣膜呈黏液性水肿性改变,瓣膜呈气球形,腱索增粗,心脏肥大和二尖瓣钙化以及皮纹异常等,本综合征的病理改变以心血管系统最显著,并具有代表性,显微镜下可见有主动脉中层弹力组织稀疏和碎裂,伴平滑肌呈不规则的螺纹状改变,胶原量增加,并可见有异染性物质呈囊状空泡散在分布于中层,主动脉夹层瘤形成者,显示为囊状中层坏死和弹力纤维中度变性,伴以平滑肌束紊乱,主动脉瓣的组织病理改变为正常结构破坏和丧失,囊状变性和组织纤维细胞丧失,皮肤的病理改变表现为有空泡状退行性变及弹力纤维排列紊乱,关节滑膜改变也是弹力纤维变性,胶原增多及异染性物质呈散在性囊状分布。
诊断依据
(1)特殊骨骼变化,即管状骨细长,尤以指,掌骨为著,骨皮质变薄,纤细,呈蜘蛛指样改变。
(2)先天性心血管异常。
(3)眼部症状。
(4)家族史。
以上临床4项标准中有3项者即可确诊,在前3项中仅出现2项改变即可诊断为不完全型马方综合征。
2.Mckusick(1995)将马方综合征的心血管异常列为
(1)主动脉扩张(升主动脉,降主动脉),主动脉夹层瘤,主动脉瓣狭窄,动脉导管未闭。
(2)肺动脉异常(肺动脉扩张,肺动脉瘤)。
(3)间隔缺损(房间隔缺损,室间隔缺损)。
(4)瓣膜异常和伴有亚急性细菌性心内膜炎。
鉴别诊断
1.与类胱氨酸尿症鉴别 本病征须与类胱氨酸尿症鉴别,后者系隐性遗传,患儿智力发育迟缓,尿含类胱氨酸。尿硝普盐试验阳性。
2.与马方综合征鉴别 此外应与反马凡综合征即Marchesanis syndrome相鉴别, 以及:①同型胱氨酸尿症;②风湿性主动脉瓣关闭不全;③家族性二尖瓣脱垂或主动脉瓣脱垂;④家族性主动脉瓣环扩张;⑤先天性挛缩性蜘蛛样指(趾)等鉴别。
小儿马方综合征西医治疗
(一)治疗
无特效治疗,主要是对症治疗。主要针对心血管畸形治疗,支持足弓和改善视力。马方综合征的药物治疗,主要目的是减缓或推迟心血管病变的发生,防治室性心律失常。
可用普萘洛尔(心得安)降低心肌收缩力、降低血流对病变主动脉的冲击,以防止主动脉壁层动脉瘤形成和破裂。一项用小鼠做的新研究提出,治疗高血压的药物洛沙坦也许对在马方综合征(Marfan syndrome)患者身上预防主动脉瘤有用。外科手术以矫正心血管畸形。对于瘤体直径大于6cm,伴主动脉瓣或二尖瓣关闭不全,出现左心室损害或心力衰竭,或合并急慢性夹层动脉瘤者,应在积极准备下尽早或急诊手术。
晶体异位轻者,可试行验光,予以矫正。若晶体脱入前房或玻璃体内而继发育光眼时,可行晶体摘除术。不能控制的心力衰竭、严重的肝肾功能衰竭以及内膜剥离所致脑神经缺血性损伤者应视为手术禁忌。
对瞳孔阻滞引起的青光眼早期可滴扩瞳剂治疗。
(二)预后
马方综合征的自然经过,尽管病变发展速度个体差异很大,但总体看来预后险恶。据Mardoch等调查,有1/3的马方综合征患者死于32岁以前,2/3死于50岁左右。1995年Sileveman报告马方综合征平均年龄仅40岁。死亡的主要原因绝大多数是心血管病变造成的。92%死于心血管并发症,主要原因是瘤体破裂、心力衰竭、房室传导阻滞、感染性心内膜炎等。手术治疗因病变组织松软,常使手术缝合发生困难,特别是换置人造瓣膜者可裂开,故手术疗效较差。
小儿马方综合征中医治疗
暂无可参考数据。
小儿马方综合征日常保健
注意饮食均衡,多吃水果、蔬菜等高纤维食物,多吃鸡蛋、大豆等高蛋白质食品,注意饮食清淡,可进行适量的运动。戒辛辣、咖啡等刺激性食物。
小儿马方综合征日常预防
预防患儿出生:
(1)婚前检查:包括详细询问男女双方及其家庭成员的健康状况既往病史及医治情况,尤其是有无先天畸形,遗传病史和近亲婚配史。应进行家系调查、血型检查染色体检查或基因诊断,以检出携带者;
(2)遗传咨询:夫妻中的一方或其亲属中有某种遗传病或先天性畸形,询问后代类似疾病的发生情况;若已作过某种遗传病或先天性畸形,询问再生育时后代情况及如何预防患儿的出生。妊娠期曾患某病、服用某药或接触有毒物质或放射线,询问胎儿的可能情况。
基础代谢率低,血清黏蛋白低于正常,尿排羟基脯氨酸增加,黏多糖增加,特别是硫酸胶质A或C增多,尿中透明质酸过多。
关于24h尿羟脯酸的测定有些学者建议作为一项诊断指标,国内同济医科大学的资料24h尿羟脯酸正常成人和小儿的结果为(24.41±17.02)mg,患者(包括成人,小儿)为(44.84±36.12)mg两者差异非常显著,本试验虽特异性较差,敏感性较低,但在除外可影响尿羟脯氨酸检查值的疾病和其他因素的前提下,若其值明显增高对诊断有意义。
1.X线检查
(1)指骨细长。
(2)掌骨指数测定,即右手第2~5掌骨长宽之比,正常为5.5~8.0;8.1~8.3提示(可能)诊断,≥8.4确诊本病。
(3)指骨指数测定,即右手环指近端指骨长宽之比,女>4.6,男>5.6,可以诊断本病。
(4)主动脉根部宽度明显扩张,主动脉逆行造影示升主动脉呈花瓶样扩张,左室增大。
2.超声心动图 国内报道诊断符合率92.3%。
(1)主动脉根部扩张:按Brown等的标准:①主动脉宽度>22mm/m2体表面积;②实测主动脉内径>37mm;③左房主动脉内径<0.7cm;具备3项中的2项者可诊断本病。
(2)主动脉瓣关闭不全征象。
(3)二尖瓣脱垂征象及二尖瓣闭锁不全征象。
(4)有主动脉夹层分离者可发现相应征象。
(5)其他心血管畸形。
3.CT和或MRI检查 可以明确发现有无主动脉病变,血管壁厚度,主动脉夹层分离及撕裂,管腔内闭塞等。
4.裂隙灯检查 可以发现晶状体脱位。
1.心血管最可能并发主动脉特发性扩张,主动脉瓣狭窄,主动脉夹层动脉瘤和二尖瓣异常等。
2.眼部病变可并发晶体脱位或半脱位,高度近视,青光眼,视网膜剥离,虹膜炎等造成视力减退甚至失明。。
3.神经系统病变可并发蛛网膜下腔出血和颈内动脉瘤,癫痫大发作,此外,马方综合征病人还可发生脊柱裂,脊柱脊髓膨出,脊髓空洞症。