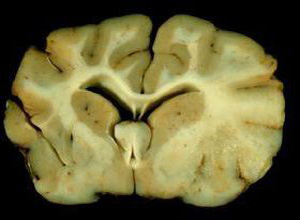
异染性脑白质营养不良(metachromatic leukoencephalopathy,MLD),亦称异染色性白质萎缩或异染色性白质脑病,1910年由Alzheimer首先报道。系常染色体隐性遗传性疾病,由芳基硫脂酶A(arylsulfatase-A)缺乏所引起。
根据起病年龄可以区分为晚期婴儿型(1~2岁起病),少年型(4~15岁起病)和成年型(16岁以后起病),以晚期婴儿型最为常见。
典型者其病程可分为以下3期:
第1期:1~2岁之间发病,病前婴儿发育正常;起病后病儿逐步出现运动减少,肌张力降低,步态蹒跚,维持姿势困难,不能独立站,坐,甚至抬头困难,体格检查可见肌张力降低,腱反射降低或消失,视盘苍白,锥体束征阴性,脑电图正常或有慢波增多,脑脊液压力正常,可有轻度蛋白质增高,此期持续数周至数个月。
第2期:病儿进行性智能减退,语言减少到消失,对周围环境逐步反应减少,尖叫而卧床不起,体格检查可见瞳孔光反应迟钝,视盘苍白萎缩,面无表情,吞咽动作缓慢,四肢肌张力增高,肢体伸直,腱反射亢进,病理锥体束征阳性,但躯干和颈肌肌张力正常或偏低,脑脊液压力,细胞正常,蛋白质明显升高,脑电图出现弥漫性慢波灶,此期病程可持续一至数年。
第3期:为晚期病症,病儿对外界极少反应,常有抽搐和肌阵挛发作,呈现特殊的去皮质强直体位,头后仰,项强直,肌强直,四肢腱反射极难引出,两侧锥体束征阳性,瞳孔散大而对光反应极差,眼球游动或是有“玩偶”眼征,吸吮和吞咽严重障碍,脑电图出现弥漫性慢波和散在的棘波,脑脊液蛋白质进一步增高,达1g/L以上,多数病儿多次继发感染,常于5~6岁左右病故。
少年型和成年型病者起病晚,进展缓慢,常有周围神经感觉缺失,晚期可有精神和行为异常。
(一)发病原因
异染色性脑白质营养不良属常染色体隐性遗传,突变基因位于第22号染色体上,由芳基硫脂酶A(aryl sulfatase-A)缺乏所引起。
(二)发病机制
硫酸脑苷脂分布于神经组织髓鞘,肾小管上皮细胞等细胞膜中,正常情况下,芳基硫酸脂酶A催化硫酸脑苷脂水解,将半乳糖硫酸脑苷脂分解为半乳糖脑苷脂和硫酸,此酶缺乏时引起硫酸脑苷脂于体内沉积。
主要的病理改变为中枢神经系统髓鞘脱失,周围神经受累轻微,病理切片中,以甲苯染色时,可见神经细胞,神经胶质细胞和巨噬细胞中有红黄色的异染物质沉积,肝,肾组织亦可同时受累。
本病临床症状与Krabbe病没有什么区别,诊断十分困难,特别是成年型患者诊断更为困难,尿,血液白细胞中芳基硫酯酶A活性降低为诊断本病的依据,病人皮肤成纤维细胞培养芳基硫酯酶A活性降低更为敏感,周围神经活检,直肠黏膜活组织检查中发现异染色性类脂质颗粒可确诊本病。
需与Pick病,Alzheimer病等鉴别。
异染性脑白质营养不良西医治疗
(一)治疗
无特效治疗。曾有人应用牛脑提取的芳基硫脂酶A 1000万U静脉或鞘内注射,虽然在治疗以后肝脏组织中酶的活性恢复正常,但脑内酶活性和脱髓鞘病变仍无任何改善。
(二)预后
多数患儿多次继发感染而于5~6岁病故。少年型和成年型患者进展缓慢,常有周围神经感觉缺失。晚期可有精神和行为异常。
异染性脑白质营养不良中医治疗
当前疾病暂无相关疗法。
平时多注意补充一定的维生素,尽量做一些患者顺口的饭菜,其他的可听取医生的建议。
进行遗传咨询,预防措施包括避免近亲结婚,携带者基因检测及产前诊断和选择性人工流产等,防止患儿出生。
尿,血液白细胞中芳基硫酯酶A活性降低。
周围神经活检,直肠黏膜活组织检查中发现异染色性类脂质颗粒。
随病情发展,出现的症状体征可以是本病表现,也可以看作本病不同类型的并发症。