慢性粒细胞性白血病是一种骨髓增殖性疾病,其特点是粒系(包括已成熟的和幼稚阶段的粒细胞)产生过多。在疾病早期,这些细胞尚具有分化的能力,且骨髓功能是正常的。本病常于数年内保持稳定,最后转变为恶性程度更高的疾病,本病患者以年龄在30~40岁间居多,20岁以下者罕见。
-
挂什么科:内科 血液科
-
需做检查:过氧化酶染色 维生素B1 维生素B12 血清钴(Co) 血清磷酸已糖异构酶 血清酸性磷酸酶同工酶 骨髓象分析 红细胞滤过指数(IF) 中性杆状核粒细胞
-
治疗方法:药物治疗 支持性治疗
-
常用药物:暂无相关信息
-
一般费用:根据不同医院,收费标准不一致,市三甲医院约(50000——100000元)
-
传染性:无传染性
-
治愈周期:1-2年
-
治愈率:30%
-
患病比例:0.006%
-
好发人群:无特定的人群
-
相关症状:
-
相关疾病:
慢性白血病在小儿时期较少,约占儿童白血病的3%~5%,其中主要为慢性粒细胞白血病(chronic myelogenors leukemia,CML)。
CML在婴儿时期的临床表现与成人CML有显著差别,故一般将小儿CML分为幼年型和成人型,文献中亦有分为婴儿型,家族型,幼儿型,成人型四型者,其中家族型与婴儿型的表现相似,只是常在近亲中发病。
1.幼年型慢性粒细胞白血病
此型几乎皆发生在5岁以下儿童,尤以2岁以下的婴幼儿多见,男性发病多于女性,可发生于家族性神经纤维瘤,生殖泌尿系畸形或智力低下的患儿。
起病可急或缓,常以呼吸道症状为主诉,多见面部斑丘疹或湿疹样皮疹,甚至为化脓性皮疹,亦可见皮肤咖啡斑,皮肤症状可出现于白血病细胞浸润前数月,淋巴结肿大,甚至为化脓性,进行性肝脾肿大,由于血小板减少而继发出血亦非罕见。
JCML起源于多能造血干细胞,故可造成红系增生障碍,血小板数与量异常,以及淋巴细胞功能异常,与成人型不同,其异常增生主要在粒单系统,体外干细胞培养主要形成CFU-GM,染色体检查多为正常,个别可见-7,+8(8三体)或+21(21三体)。
周围血象白细胞增高,血小板减低和中度贫血,白细胞中度增高,多在100×109/L以下,未成熟粒细胞和有核红细胞可出现在周围血中,并有单核细胞增多,白细胞碱性磷酸酶降低,偶正常,血清和尿中溶菌酶增高,HbF增高,骨髓粒:红为3~5:1,粒系和单核系增生旺盛,可见红系增生异常,原始粒细胞在20%以下,巨核细胞减少,体外骨髓细胞培养以单核细胞为主。
由于JCML常有发烧,肝脾肿大,中度贫血,白细胞增多,需与感染所致的类白血病反应鉴别,还应与传染性单核细胞增多症鉴别。
2.成人型慢性粒细胞白血病
发病年龄在5岁以上,以10~14岁较多见,很少见于3岁以下儿童,男女差别不大,由于是多能造血干细胞的恶性增殖,故粒系,红系,巨核系等多系受累,急变期可转变为淋巴细胞白血病,约85%以上的患儿存在Ph1染色体(即t(9:22)),对Ph1染色体阴性者,用分子生物学技术又可分为有bcr重组(phbcr+CML)和无bcr重组(PH-bcr-cml)两亚型,前者临床症状与PH1染色体阳性者类似,后者临床症状不典型。
发病缓慢,开始时症状较轻,表现为乏力,体重减轻,骨关节疼痛,体征可见巨脾,肝脏肿大,淋巴结轻度肿大,视神经乳头水肿等,很少有出血症状。
周围血象主要为白细胞增多,80%在100×109/L以上,血色素在80g/L左右,血小板增多,分类可见粒系增多,包括嗜酸,嗜碱粒细胞增多,原始粒细胞增多不明显,以中,晚幼和成熟粒细胞为主,白细胞碱性磷酸酶减低,HbF不增高,血清免疫球蛋白不增高,骨髓增生活跃,以粒系增生为主,原始粒细胞<10%,多为中,晚幼粒细胞及杆状核细胞,粒:红为10~50:1,部分患者可见骨髓纤维化,骨髓巨核细胞明显增多,以成熟巨核细胞为主,血清和尿溶菌酶不增高,但VitB12和VitB12运载蛋白增高,骨髓培养集落与丛落皆增多。
(一)发病原因
离子辐射(25%):离子辐射可以使CML发生率增高,在广岛和长畸原子弹爆炸后幸存者、接受脊椎放疗的强直性脊椎炎患者和接受放疗的宫颈癌患者中CML发病率与其他人群相比明显增高。
长期接触苯或进行化疗(20%):长期接触苯和接受化疗的各种肿瘤患者可导致CML发生,提示某些化学物质亦与CML发关。CML患者HLA抗原CW3和CW4频率增高,表明其可能是CML的易感基因。
其他(5%):尽管有家族性CML的报道,但CML家族性聚集非常罕见,此外单合子双胞胎的其他成员CML发病无增高趋势,CML患者的父母及子女均无CML特征性Ph染色体,说明CML是一种获得性白血病,与遗传因素无关。
(二)发病机制
1、起源于造血干细胞:CML是一种起源于造血干细胞的获得性克隆性疾病,其主要证据有:
①CML慢性期可有红细胞、中性粒细胞、嗜酸/嗜碱粒细胞、单核细胞和血小板增多。
②CML患者的红系细胞、中性粒细胞、嗜酸/嗜碱粒细胞、巨噬细胞和巨核细胞均有Ph染色体。
③在G-6-PD杂合子女性CML患者中,红细胞、中性粒细胞、嗜酸/嗜碱粒细胞、单核细胞和血小板表达同一种G-6-PD同工酶,而成纤维细胞或其他体细胞则可检测到两种G-6-PD同工酶。
④每个被分析的细胞其9或22号染色体结构异常都一致。
⑤分子生物学研究22号染色体断裂点变异仅存在于不同CML患者,而在同一病人的不同细胞中其断裂点是一致的。
⑥应用X-连锁基因位点多态性及灭活式样分析亦证实了CML为单克隆造血。
2、祖细胞功能异常:相对成熟的髓系祖细胞存在有明显的细胞动力学异常,裂指数低、处于DNA合成期的细胞少,细胞周期延长、核浆发育不平衡,成熟粒细胞半衰期比正常粒细胞延长。采用3H自杀试验证实仅只有20%的CML集落处于DNA合成期,而正常人为40%,CML原粒、早幼粒细胞标记指数比正常人低,而中、晚幼粒细胞标记指数与正常对照相比无明显差别。造血祖细胞集落培养发现CML骨髓祖细胞与外周血祖细胞的增殖能力不同,骨髓CFU-GM和BFU-E数与正常对照相比通常增高,但亦可正常或减低,而外周血可升高至正常对照的100倍。Ph阳性CML病人骨髓细胞长期培养研究发现,经几周培养后在培养基中可检测到Ph阴性的祖细胞,现已证实这主要为CML造血祖细胞黏附功能异常所致。
3、分子病理学:1960年,Nowell和Hungerfor描述了CML相关的Ph染色体,这是首次发现的与一特异人类肿瘤相关的非随机染色体异常。1973年Rowley采用奎宁和姬姆萨染色技术首次证实CML中发现的Ph染色体(22q-异常)是t(9;22)(q34;q11)染色体易位所致。1982年在9q34断裂区克隆出了ABL基因。1983年证实位于q34的基因片段易位到22号染色体上与22q11断裂区一个称为BCR的基因形成BCR-ABL融合基因。
(1)ABL基因:原癌基因c-abl定位于q34,在物种发育过程中高度保守,编码在所有哺乳动物组织和各种类型细胞中均普遍表达的一个蛋白质,c-abl长约230kb,含有11个外显子,走向为5′端至着丝粒。该基因第一个外显子有两种形式,外显子1a和1b,因而有两种不同的c-abl mRNA,第一种称为1a-11,长6kb,包括外显子1a-11。另一种称为1b,自外显子1b开始、跨越外显子1a和第一个内含子,同外显子2-11相接,长为6kb,这两种ABL的RNA转录编码两种不同的分子量均为145000的ABL蛋白。DNA序列分析发现。c-abl属非受体蛋白-酪氨酸激酶家族,除激酶 片断外,该基因还有在信号传导蛋白的相互作用和调节中非常重要的SH2和SH3片断,c-abl的特征是有一个大的C末端非催化片断,该片断含有DNA和细胞骨架结合的重要序列和一个参与该传导信号的区域。正常的p145ABL穿梭于细胞核和胞浆之间,主要定位于细胞核,具有较低的酪氨酸激酶活性。p145ABL的活性和细胞内定位受连接细胞骨架与细胞外间质的整合素(integrins)调控,现有研究表明至少在纤维细胞,ABL激活需要细胞黏附,因此ABL可能通过将整合素信号传递至细胞核从而充当黏附和细胞周期信号之间的桥梁,参与细胞生长和分化控制。
(2)BCR基因:BCR基因定位于22q11,长130kb,有21个外显子,起始方向5′端至中心粒。有4、5kb和6、7kb两种不同的BCR mRNA转录方式,编码一分子量为160000的蛋白p160 BCR,该蛋白有激酶活性。p160 BCR之C末端与ras相关的GTP结合蛋白p21的GTP活性有关联。
(3)BCR-ABL基因:位于9q34的c-abl基因易位于22号染色体与位于22q11的bcr基因形成BCR-ABL融合基因。迄今CML患者中已发现有3个bcr断裂点丛集区,分别为M-bcr、m-bcr、u-bcl和6种BCR-ABL融合转录方式,与M-bcr相应的有b2a2、b3a2、b2a3,其编码蛋白为p210,与m-bcr相应的有ela2,其编码蛋白为p190,与u-bcr相应的有e19a2,其编码蛋白为p230。
小鼠模型体内已证实BCR-ABL可导致CML发生,BCR-ABL融合蛋白定位于细胞浆,具有极高的酪氨酸激酶活性,通过改变作为BCR-ABL催化底物的一些关键的调节蛋白磷酸化状况激活多种信号传导途径,如通过激活参与细胞增殖和分化调控的Ras信号途径,使祖细胞数量增多,干细胞池减少,干细胞成为增殖池的一部分,从而使未成熟粒细胞不断扩增。BCR-ABL作用的另一种机制是改变正常整合素功能,正常造血祖细胞黏附于细胞外基质,而黏附是由祖细胞细胞表面受体特别是整合素来介导的,BCR-ABL通过干扰β1整合素的功能导致CML细胞的细胞黏附功能缺陷,从而使未成熟细胞释放至外周血并迁移至髓外部位。
最近,CML发病机制研究又取得了进展:①体外培养发现,BCR-ABL通过抑制凋亡而延长CML祖细胞的因子非依赖性生长时间。②用反义寡核苷酸下调BCR-ABL表达后可能通过增加细胞对凋亡的敏感性从而抑制白血病细胞在小鼠体内生长,特别是减少CML病人早期祖细胞集落形成,降低CML样细胞系的细胞增殖。③表达BCR-ABL的、转化的、因子非依赖性的、可致瘤的小鼠造血细胞通过上调bcl-2而增加对凋亡的敏感性,当bcl-2表达受抑后,BCR-ABL阳性细胞又变成了因子依赖性和非致瘤性。以上实验结果表明,BCR-ABL抑制细胞凋亡导致髓系细胞不断扩增是CML的又一发病机制。
(4)急变发生机制:细胞遗传学研究发现80%的AP或BP CML患者有继发性染色体异常,最常见的异常依次为+8、+Ph、i(17)、+19、+21和-Y。急性粒细胞白血病变(急粒变)的患者中约80%有非随机性染色体异常,其染色体核型常表现为超二倍体,最常见的异常为+8,且+8常与其他染色体异常如i(17)、+Ph、+19等同时出现,其次为+Ph、i(17)和-Y。急性淋巴细胞白血病变(急淋变)的患者约30%有继发性克隆性染色体异常,常为染色体丢失,从而表现为亚二倍体或结构异常,常见异常为+Ph和-Y,+8少见,i(17)尚未见报道,-7、14q+与急淋变特异相关。尽管有研究发现急变期CML有N-Ras基因突变和c-Myc基因表达增高,但其发生率极低。Rb基因在急变期CML患者亦极少有改变。Sill等发现p161NK4A基因的纯合子缺失与CML急淋变相关。CML急性变分子机制研究较多的还是p53基因,20%~30%的急粒变的患者存在有p53基因结构和表达的异常,CMLp53基因改变特征为:①主要改变是基因重排和突变。②主要见于急粒变,急淋变极少见。③p53突变常见于有17P-异常患者。④p53突变能导致CML的急粒变。最近,又有钙调素基因甲基化程度、端粒长度和端粒酶活性改变与CML急变关系的研究报道,但其意义尚待进一步阐明。
诊断
根据病史、临床症状和实验室检查资料可以诊断。 鉴别诊断
1、其他原因引起的脾大:血吸虫病,慢性疟疾,黑热病,肝硬化,脾功能亢进等均有脾大,但各病均有各自原发病的临床特点,并且血象及骨髓象无CML的改变,Ph染色体阴性。
2、类白血病反应: 常继发于严重感染,恶性肿瘤等疾病,并有相应原发病的临床表现,白细胞数可达50×l09/L,粒细胞胞浆中常有中毒颗粒和空泡,嗜酸性粒细胞和嗜碱性粒细胞不增多,NAP反应强阳性,Ph染色体阴性,血小板和血红蛋白大多正常,原发病控制后,类白血病反应亦随之消失。
3、骨髓纤维化: 原发性骨髓纤维化症脾大显著,血象中白细胞增多,并出现幼粒细胞等,易与CML混淆,但原发性骨髓纤维化症的外周血白细胞数一般比CML少,多不超过30×l09/L,且波动不大,NAP阳性,此外幼红细胞持续出现于外周血中,红细胞形态异常,特别是泪滴形红细胞易见,Ph染色体阴性,多次多部位骨髓穿刺干抽,骨髓活检网状纤维染色阳性。
慢性粒细胞性白血病西医治疗
1、治疗原则:
对于慢粒的治疗不必操之过急,白细胞计数在100×109/L以下的患者不需立刻治疗、因为循环中主要是成熟的粒细胞,其体积较原始细胞小且具有较好的变形能力,白细胞计数在200×109/L以上者需采取积极治疗措施。当前以采用细胞毒药物作化疗为主。对于那些因白细胞极度增生而出现的症状,如有阴茎异常勃起、呼吸窘迫、视力模糊、心理变态等,则应在进行急性的白细胞除去术的基础上联用骨髓抑制剂进行治疗。
2、化学治疗:
有效的药物有BUS(马利兰)、HU(羟基脲)、CTX、CLB、6-MP(6-巯基嘌呤)、MMC(丝裂霉素)。其中以BUS为首选药物,其次为HU。BUS是目前最有效的药物,缓解率在95%以上,服用方便为此药之优点。用法为2mg每日3次,一直用至白细胞降至14×109/L以下停用或间歇给药。一般规律是用药1~2周自觉症状好转,4~6周明显好转。当白细胞减至10×109/L时,减量至1~2mg/d,一直维持2~3个月。停药后,如白细胞波动在10~50×109/L间,可考虑小剂量维持1年以上。白细胞减少到5~10×109/L血小板在100×109/L以下,或者有慢粒急变倾向才应停药。马利兰的毒副作用主要是骨髓抑制,特别是血小板减少。个别病人虽用药量不大也会出现全血细胞减少,恢复较慢。长期服用此药可引起肺纤维化,皮肤色素沉着。类似慢性肾上腺皮质功能减退的症状,精液缺乏或停经。
HU开始剂量为每日3g,口服。用后白细胞数下降很快。当降至20×109/L左右时,将剂量减至一半;降至10×109/L时,将剂量再减少。维持剂量约每日0.5~1.0g。一般不完全停药,因停药后白细胞计数很快上升。此药优点是作用快;如果白细胞下降过多,停药后能很快上升;副作用少。缺点是需经常验血以指导治疗。另外,亦可联合α-IFN(α-干扰素)治疗慢粒。
方法,口服HU2.0~6.0g/d,同时皮下注射。α-IFV300万u,iv,每周3次,应用8~32周。当白细胞降至10×109/L,HU减少继续用1~2周,根据情况停用或用小剂量。HU维持量为0.5~1.0/d,有条件者可继续用。α-IFN300万u,iv,每周一次。用药期间每周查血常规2次,骨髓象每4周检查一次。
3、放射治疗:
深部x线,用深部x线对全身和局部的肝脾区以及浸润部位照射。脾区照射开始剂量为50cGy,以后每日或隔日100~200cGy。白细胞降至20×109/L时停止。对化疗效果不佳或复发的可以用放疗,据报道,其疗效不低于BUS。核素32P治疗,仅用于对BUS及脾区放疗效果不佳者。32P剂量是根据白细胞增多程度而定,若白细胞总数>50×109/L,32P的开始剂量为1~2.5mCi,静注。2周后再用1~1.5mCi,以后每隔2周给同样剂量1次,待白细胞降至20×109/L时停用。在缓解期间,每1~3个月观察1次,当白细胞>25×l09/L时,可再给1~1.5mCi。
4、脾切除术:
脾脏可能是慢粒急变的首发部位,切除脾脏可能延缓急变和延长患者存活期。切除脾脏的手术指征:
①确诊为慢粒者。
②对化疗反应良好。
③65岁以下且无大手术禁忌症者。慢粒急变是手术的禁忌症。
5、骨髓移植:
年龄在45~50岁在慢性期的病人,以亲兄弟姐妹HLA相同的异基因骨髓作移植。移植成功者,一般能获得长期的生存或治愈。
6、其他治疗:
化疗前如果白细胞数在500×109/L以上,可先用血细胞分离机作白细胞除去术以迅速降低白细胞数,避免白细胞过多可能阻塞微血管而引起的脑血管意外的危险。化疗开始时,特别是用Hu治疗时,宜同时加用别嘌呤醇0.1g每日3次,以防止细胞破坏过多过速而引起尿酸肾症。
7、慢粒急变的治疗:
慢粒急变的治疗比急性白血病的治疗困难,完全缓解仅10.7%,目前慢粒急变的治疗方案如下:Ara-c(环阿糖胞苷)100mg/m2·d,第1~14天;ADM(阿霉素)30mg/m2·d,第1~3天;VCR2mg,第1天;上述药物相继静脉输注。PDN40mg/m2·d,分次口服,第l~7天。
慢性粒细胞性白血病中医治疗
一、辨证施治
(1)气滞血瘀
治法:疏肝理气,活血化瘀。
方药:膈下逐瘀汤加减。方中延胡索、乌药、枳壳疏肝理气;红花、当归、莪术、三棱、五灵脂活血化瘀;丹皮、赤芍具有活血止血双重作用,以防活血过量引起出血。可佐以青黛、 雄黄解毒祛瘀;甘草调和诸药。如纳差加砂仁。
(2)正虚瘀结
治法,益气养血,活血散瘀。
方药:八珍汤加味。八珍汤为益气养血之代表方,方中党参、白术、茯苓,健脾益气;当归、首乌、阿胶补血;三棱、莪术、红花、延胡索活血散瘀;佐以青黛、雄黄解毒祛瘀。如阴血不足,加地黄、麦冬、枸杞子滋阴,出汗多加用五味子敛阴。
(3)热毒炽盛
治法:清热凉血。
方药:犀角地黄汤或清营汤加减。方中犀角(以水牛角代替)、生地、丹皮、赤芍、清热凉血;金银花、黄芩、黄连清热解毒,佐以青黛、白花蛇舌草、龙葵以解毒。壮热不退加生石膏、知母、生甘草。便血加用白芨粉、三七粉;尿血选大蓟、小蓟;齿龈出血加藕节、白茅根等。
二、中成药
(1)当归龙荟丸:6~10g,日2~3次。
(2)青黛:分装胶囊,每次2~4g,每日3次,饭后吞服。
(3)青黄散:青黛9份,雄黄1份,混匀装胶囊,每次3~5g,一日3次,饭后服,维持量3~6g/日,服药1~3个月后排砷1次,用二巯基丁二钠1.0 溶于40ml生理盐水中缓慢静脉注射,连用3天。
(4)六神丸:每次30粒,一日3次口服。
(5)梅花点蛇丹:每次10粒,一日3次口服。
(6)牛黄解毒片:每次3~4片,一日3次。
(7)大黄zhe??虫丸:每次1丸,一日2~3次。
(以上提供资料及其内容仅供参考,详细需要咨询医生。)
食疗方(仅供参考,详细请询问医生)
(1)鹅血粥 :鹅血100克,粳米50克。将新鲜鹅血隔水蒸熟备用,粳米煮熟,加人鹅血、食盐煮开饮服,常服有效。
(2)粳米、猪肝、莲子、大枣粥 :粳米50克,莲子20克(水泡),熟猪肝(切成丁)30克,大枣10个,加水适量熬粥,早晚分服,有防治贫血的作用。
(3)大枣桂圆薏米粥 :大枣10个,桂圆20克,薏米40克,加水适量熬成粥,早晚食用。大枣、桂圆、慧米均为健脾益胃滋补之品,经常食用可增强体质,提高机体抗癌免疫功能。
慢粒患者饮食应遵循6宜7忌
【宜】
(1)肝脾肿大宜吃赤豆、李、大枣、裙带菜、甲鱼、龟、海带、紫菜。
(2)出血宜吃藕、葡萄、荠菜、蘑菇、香菇、木耳、金针菜、猫肉、鲛鱼。
(3)贫血宜吃猪肝、黄鱼、海参、鲩鱼、鲵鱼、香蕈、芝麻、蜂乳。
(4)宜多吃具有抗白血病作用的食物:蟾蜍、苜蓿、蒜、小麦、胡萝卜、核桃、蒲公英、牡蛎。
(5)发热宜吃豆豉、葱白、冬菜、蕹菜、李、银杏、绿豆、苦瓜、菱、节瓜、海鳗、鳖鱼、猪脊髓。
(6)淋巴结肿大宜吃芋艿、栗、桑椹、核桃、荔枝、荸荠、黄颡鱼、猫肉、羊肚、鲎、文蛤、牡蛎、龟、甲鱼。
【忌】
(1)忌咖啡、浓茶等兴奋性饮料。
(2)忌羊肉、狗肉、韭菜、胡椒等温热性食物。
(3)忌公鸡、猪面肉等发物。
(4)忌猪脚、鸡内脏及头脚、蟹、鲤鱼、鲫鱼等。
(5)忌烟、酒。
(6)忌葱、蒜、姜、桂等刺激食品。
(7)忌肥腻、油煎、霉变、腌制食物。
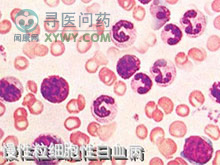
1、血象:白细胞计数高在100×109/L以上,血片中大多为中性杆状核和晚幼粒细胞,其余为分叶核,中幼粒,早幼粒和少数原始粒细胞,嗜酸性及嗜碱性粒细胞亦增多,早期血红蛋白及红细胞轻度减少,血小板正常或增加,晚期红细胞和血小板减少,在血象方面须与类白血病反应相鉴别。
2、骨髓象:骨髓呈增生明显至极度活跃,细胞分类与周围血相似,骨髓片中,可见到各期粒细胞,其中以中,晚幼粒为主,原粒细胞及早幼粒较正常增多,但一般不超过5%~10%,嗜酸和(或)嗜碱性粒细胞增多,红细胞系相对减少,粒:红约10~50:1,幼红细胞和巨核细胞早期常增多,晚期减少,90%患者成熟的中性粒细胞碱性磷酸酶活性明显降低。
3、染色体检查:Ph’染色体见于90%以上慢粒病人,Ph’染色体被认为是慢粒多能干细胞的肿瘤性标志,少数慢粒病人Ph’染色体为阴性,根据有无Ph’染色体可将慢粒为Ph’阳性和Ph’阴性两大类,前者预后优于后者。
4、血液生化:血清维生素B12浓度及维生素B12结合力显著增高为本病特点之一,增高的幅度与白细胞增多程度成正比,增高的原因是大量正常的和白血病性粒细胞产生了过多运输维生素B12的转钴蛋白I,血清尿酸浓度可以增高,尤其在化疗时。
当白细胞计数>100×109/L时,可有白细胞淤滞综合征,出现呼吸困难,紫绀,脏器梗死,眼底静脉扩张,视神经乳头水肿,眼底出血,神经改变甚至中枢神经系统出血等表现,巨脾者可伴脾脏梗死。