
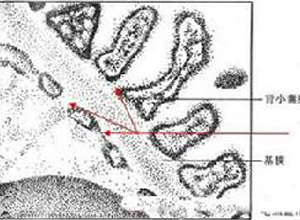
肾小球基底膜蛾噬现象是指甲-髌骨综合症的诊断依据之一。对肾活检标本进行判断不能仅用肾小球基底膜蛾噬现象,必须用磷钨酸染色鉴定原纤维因其敏感性更高,对诊断更有价值。
指甲-髌骨综合征为遗传性疾病其传递方式是常染色体显性遗传。发生率为4.5/100万~22/100万,没有性别的差异。患者传递给其后代的机会为50%,基因位点与腺苷酸环化酶和ABO血型的位点连锁位于9号染色体上。Looij等根据自己的数据和资料推断,如果本综合征家族的一个患者有明显的临床肾脏表现,则其所生孩子患肾病的危险性是1/4,发展至肾功能衰竭的概率是1/10。
病因:本综合征是常染色体显性遗传,基因位点与腺苷酸环化酶和ABO血型的位点连锁位于9号染色体上。
发病机制:对本综合征发病机制了解甚少,有人认为是一种胶原疾病,在胶原蛋白的合成、装配或降解过程中存在异常。对这种疾病的细胞学机制尚未研究。病理改变缺少非肾小球性基底膜损害这一点提示本综合征各种损害可能源于不同机制,并非所有损害都与基底膜异常有关。少数患者发展为抗肾小球基底膜肾炎,支持肾小球基底膜组分异常这一假说。使用针对古德帕斯丘(Goodpasture)表位的单克隆抗体的研究结果发现,2/3患者肾活检标本的肾小球基底膜不与该单克隆抗体结合,提示这一综合征的基底膜组分存在某种程度的异质性,还提示存在古德帕斯丘(Goodpsture)抗原缺失或改变。值得指出的是,尚不清楚这是本综合征原发的还是继发的改变。
指甲-髌骨综合征的主要诊断依据是家族史,典型的临床表现是骨骼的X线征和蛋白尿。必要时进行肾活组织检查。
临床上多见于青少年,其肾脏损害的主要表现为蛋白尿、镜下血尿水肿及高血压,偶见肾病综合征,病程相对良性,仅10%的病人晚期进入肾衰竭。肾外表现为指甲营养障碍、一侧或双侧髌骨缺如、肘关节畸形、角型骨盆及其他骨骼异常。指甲-髌骨综合征多数因髌骨缺如导致行走困难引起注意,根据典型骨骼改变多可诊断,伴有肾脏损害则多可确诊。放射检查显示髂骨角为特征性改变,有明确诊断意义。
有报道少数患者有肾小球基底膜超微结构改变而无骨骼、皮肤、指(趾)甲、及本综合征其他典型表现,这些患者被认为是该综合征的顿挫型或单一肾病变异类型。但这些研究所发表的电子显微镜照片并不能有力地支持这一观点。
对肾活检标本进行判断不能仅用肾小球基底膜蛾噬现象,必须用磷钨酸染色鉴定原纤维因其敏感性更高,对诊断更有价值。
治疗:
对于指甲-髌骨综合征的肾损害无特异治疗方法,一般治疗同其他肾脏疾病。对于那些进展至终末期肾脏病的患者可进行透析或肾移植一般移植的肾脏无复发的基底膜损害。
预后:有关本综合征长期预后的研究论文很少,现有报道显示10%的患者发展至终末期肾病。指甲-髌骨综合征多数预后良好。
预防:指甲-髌骨综合征系遗传性疾病,无有效预防措施。