
皮肤淋巴肉芽肿病(cutis lymphogranulomatosis)大多数报告的皮肤霍奇金病,实际上是A型淋巴瘤样丘疹病。同义名Hodgkin淋巴瘤(Hodgkin lymphoma),Hodgkin肉芽肿(Hodgkin granuloma),Hodgkin副肉芽肿(Hodgkin paragranuloma),Hodgkin肉瘤(Hodgkin sarcoma),恶性淋巴肉芽肿病(lymphogranulomatosis maligna)。本病是ML的一种,具有特殊的瘤巨细胞(Reed-Sternberg细胞,简称R-S细胞),常有不同程度的炎症细胞浸润。此二病有相当多的重叠表现。淋巴瘤样丘疹病的A型细胞与Reed-Sternberg细胞(RS细胞)有共同的免疫表型标志。淋巴瘤样丘疹病可见于患霍奇金病的患者。原发性皮肤霍奇金病而无淋巴结受累者很难证实而且是十分罕见的。
(一)发病原因
本病病因尚不清楚。有下列一些看法:
1.病毒感染:与患者密切接触的人发病率高,Lerine等提出患者血清中EB病毒抗体效价增高,认为与EB病毒感染有关,但尚无确切证据。
2.射线影响:原子弹爆炸区居民的发病率较其他区居民高4倍。
4.细胞免疫缺陷:根据患者血中T淋巴细胞减少、细胞免疫低下、正常迟发型变态反应消失、对同种异体移植排斥缓慢、早期病变常从淋巴结T区开始发生,认为与T细胞免疫监视功能缺陷而导致“网状细胞”异常瘤性增生。
(二)发病机制
发病机制还不清楚。由于对R-S细胞的起源尚有争论,有待进一步探讨。对R-S细胞的起源,有认为来自T淋巴细胞、B淋巴细胞、组织细胞或交指状网状细胞或树突状网状细胞等看法。目前大多倾向于来自网状细胞,但尚未证实。
患者常诉浅表淋巴结或淋巴结群特别是在颈部呈无痛性进行性肿大,伴乏力、食欲不振、消瘦、不规则低热、盗汗和瘙痒。晚期可累及任何器官。因颈和腋部淋巴结肿大,可压迫臂神经丛而引起咳嗽、呼吸困难、上腔静脉综合征、言语困难等症状。脾脏受累可引起腹部不适、消化不良、恶心、呕吐和因脾功能亢进所产生症状。累及骨骼者可引起骨折、突眼、脊髓受压和瘫痪。中枢神经系统受累可致偏瘫、周围神经病变和疼痛。胃肠道受累可引起溃疡、疼痛、出血、梗阻和吸收不良综合征。累及肝脏可引起黄疸和肝性昏迷。泌尿生殖系统受累可引起尿毒症。患者因细胞免疫低下,易招致病毒、细菌和念珠菌感染。
皮肤损害可分为特异性和非特异性。
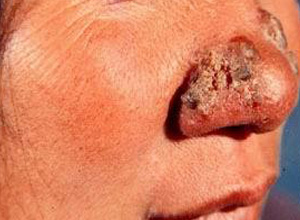
1.特异性损害:据Benninghoff等统计,见于5%~10%患者。原发性少见。大多来源于血源播散,常见于有广泛淋巴结病变的晚期患者;若由相应淋巴结逆行传播,可见于早期。原发性损害可先于其他症状,包括淋巴结肿大在数个月或5~10年出现,预后较佳,患者可存活10~20年。发生继发性损害的患者常于数个月内死亡。损害好发于头皮、颈部和躯干上半部,可表现为丘疹、结节、斑块、红皮病或湿疹样,偶或皮下结节。溃疡可在结节或斑块的基础上发生,或因淋巴结或骨骼等病变波及皮肤所致。
2.非特异性损害:较常见,常表现为表皮剥蚀、红斑、苔藓样变、痒疹样或红皮病等。其他如皮肤苍白、荨麻疹、多形红斑、结节红斑、毛发红糠疹、类天疱疮、鱼鳞病、皮肌炎、皮肤异色病样等损害也可发生。有时还可表现紫癜、大疱、淋巴水肿、秃发、掌(跖)角化以及静脉炎和色素沉着等。由于细胞免疫功能低下,至少约15%患者发生带状疱疹。
大多数霍奇金病通常起源于淋巴结,从淋巴结或经淋巴管逆行扩展至皮肤或直接扩展至皮肤。皮肤损害呈丘疹或结节,伴发溃疡或不伴发。晚期病例可发生皮肤粟粒状播散。霍奇金病患者常见非特异性皮肤损害,泛发性严重瘙痒可发生于本病其他症状出现数月数年前或本病已明确诊断之后。可因搔抓而发生继发性结节性痒疹和色素沉着。其他皮肤表现如获得性鱼鳞病,剥脱性皮炎和泛发性严重的带状疱疹。
非特异性皮肤损害不论在临床或组织病理上均无特殊性。特异性皮肤损害的临床形态无特殊性,主要靠组织病理检查明确诊断。
常见的几种肉芽肿:
性病性淋巴肉芽肿,又可称为腹股沟淋巴肉芽肿,是由沙眼衣原体感染引起的性传播疾病。本病通常在世界上热带与亚热带地区出现,尤其以南美、非洲及东南亚多见,我国较少发病。
嗜酸性肉芽肿(EG)是一种孤立性的组织细胞的非肿瘤性质的异常分化。嗜酸性肉芽肿是郎罕氏细胞增多症的一种表现,以前称为组织细胞增多症X。嗜酸性肉芽肿多发生于5-10岁的儿童,侵犯部位为骨骼和肺。这占郎罕氏细胞增多症病例的60-80%。黑人本症极少见。
非嗜酸性肉芽肿性前列腺炎镜下为非干酪肉芽肿,其中央液化坏死,近腺体管腔中有主要的组织细胞增多,周围绕以淋巴细胞、浆细胞和嗜伊红细胞。
蕈样内芽肿过去被误称为蕈样酶菌病,现已证实是一种原发于皮肤辅助性T淋巴细胞的肿瘤。多发生于青壮年,但亦有10余岁或60余岁发病,男性多于女性。有三种类型,其中最常见的是由斑片或斑块发展成肿瘤,也可直接发生肿瘤,称突发性蕈样肉芽肿,发展较快,预后较差。约10%的病例可发生红皮病,称为红皮病型蕈样肉芽肿。本病可持续数月至数年,平均为4-10年。
环状肉芽肿(granuloma annulare)是一种病因不明的良性炎症性皮肤损害,发生于真皮或皮下组织,以环状丘疹或结节性皮损为特征。
患者常诉浅表淋巴结或淋巴结群特别是在颈部呈无痛性进行性肿大,伴乏力、食欲不振、消瘦、不规则低热、盗汗和瘙痒。晚期可累及任何器官。因颈和腋部淋巴结肿大,可压迫臂神经丛而引起咳嗽、呼吸困难、上腔静脉综合征、言语困难等症状。脾脏受累可引起腹部不适、消化不良、恶心、呕吐和因脾功能亢进所产生症状。累及骨骼者可引起骨折、突眼、脊髓受压和瘫痪。中枢神经系统受累可致偏瘫、周围神经病变和疼痛。胃肠道受累可引起溃疡、疼痛、出血、梗阻和吸收不良综合征。累及肝脏可引起黄疸和肝性昏迷。泌尿生殖系统受累可引起尿毒症。患者因细胞免疫低下,易招致病毒、细菌和念珠菌感染。
(一)治疗。限局性损害X线放射治疗。亦可试用PUVA疗法。晚期患者常用联合化疗。
(二)预防。原发性损害可先于其他症状,包括淋巴结肿大在数个月或5~10年出现,预后较佳,患者可存活10~20年。