珠蛋白生成障碍性贫血(thalassemia)原称地中海贫血(mediterranean anemia),是由于正常血红蛋白中一种或几种珠蛋白肽链合成障碍或完全抑制为特征的异常血红蛋白病,属较常见的常染色体不完全显性遗传性溶血性贫血。
-
挂什么科:儿科 儿科综合
-
需做检查:蛋白电泳 尿常规 心电图 多染性红细胞 红细胞渗透脆性试验 红细胞渗透脆性试验(ROFT) 血常规 血红蛋白F包涵体生成试验 异丙醇试验 异丙醇试验 有核红细胞 胎儿血红蛋白(HbF)酸洗脱试验
-
治疗方法:药物治疗 支持性治疗
-
常用药物:暂无相关信息
-
一般费用:根据不同医院,收费标准不一致,市三甲医院约(5000 —— 8000元)
-
传染性:无传染性
-
治愈周期:1-3个月
-
治愈率:30%
-
患病比例:0.0001%
-
好发人群:儿童
-
相关症状:
-
相关疾病:
1.α-珠蛋白生成障碍性贫血
感染和(或)氧化性药物可诱发或加重溶血性黄疸,甚至“溶血危象”,类似红细胞G6PD缺陷症临床表现,应注意早知鉴别,α-珠蛋白生成障碍性贫血及其临床特征。
2.β地中海贫血
绝大多数于妊娠期30~40周(平均34周)时,胎儿死于宫内或娩出后短期内死亡,全身重度水肿,腹水,呈蛙腹,少数病例无水肿及腹水,重度贫血, 苍白 ,可有轻度黄疸,肝肿大比脾肿大明显,可无脾大,可见皮肤出血点,胎盘巨大且粗厚,苍白,质脆。
HbCS的纯合子状态:可有轻度低色素性贫血,有时发生黄疸,肝脾轻度肿大,红细胞大小不等,有靶型细胞,MCH偏低,网状红细胞计数增多,HbCS 0.05~0.06,微量HbBart’s,HbA2及F均正常,其余为HbA,这种病例很少见。
HbCS的杂合子状态(即HbCS特性):无血液学异常,或轻度贫血,红细胞异常,小红细胞症等,HbCS约0.01,HbA及A2均正常。
HbCS若同时复合α地贫1(基因型为αCSα/--)时,其临床表现和血象与HbH病相似,称为CS型HbH病,使用pH 8.6的淀粉凝胶电泳容易与HbA,HbA2,HbF等区分可来,由于量少,容易忽视。
根据β-肽链基因的缺失程度和β链合成受阻(部分或完成抑制)程度可分为以下几种类型:
依地中海贫血发病年龄,病情轻重等可分为以下3型:
(1)轻型:是正常基因和珠蛋白生成障碍性贫血βo,β ,δβ基因的杂合子状态,本型常见,儿童或青少年期发病,轻度或无贫血(Hb 100~120g/L),轻度或无肝,脾肿大,婴儿期可有轻度黄疸和脾肿大,预后良好;多在重型患者家族调查中被发现。
(2)重型:也称库理(Cooley)贫血,为βo,β 基因纯合子或双重杂合子状态,其临床特点为多数于婴儿期发病,50%在生后6个月内发病,偶见新生儿期发病者,发病年龄愈早,病情愈重,严重的慢性溶血性贫血,需依靠输血维持生命,新生儿期无贫血,HbBart’s含量25%,少量HbH,呈严重的慢性进行性贫血,血红蛋白常<30g/L;特殊面容,表现为头大,颧骨略高,鼻梁低陷,眼距增宽,表情呆钝;肝,脾日渐肿大,以脾肿大为主,可达盆腔,常并发感染,预后差,多于5岁前死于心力衰竭和感染,经治疗者常于20岁前死于心律不齐或心力衰竭。
(3)中间型:慢性溶血性黄疸型,是β 基因纯合子状态(高F)或δβo珠蛋白生成障碍性贫血,其临床表现介于重型与轻型之间,本型少见,临床特点为发病年龄较晚(常于4~5岁);中度贫血;肝轻度肿大,脾轻度到中度肿大,常有持续性轻至中度黄疸;红细胞形态与重型类似;HbF含量40%~70%,HbA2含量正常或降低;不需治疗,可活至成人期,感染和(或)药物加重溶血,可合并胆石,高间接胆红素血症,切脾后黄疸不消退。
(一)发病原因
人类各种血红蛋白的合成是由其相应的珠蛋白基因专一性控制,正常人血红蛋白(Hb)中的珠蛋白有4种肽链,即α,β,γ和δ,根据珠蛋白肽链组合的不同而形成3种血红蛋白,即HbA(α2β2),HbA2(α2δ2)和HbF(α2γ2),HbA是成人红细胞中的主要血红蛋白,约占Hb中的95%,HbA2约占成人Hb中的2%,HbF是胎儿期及早期新生儿期的主要血红蛋白,刚出生时约占新生儿Hb中的70%,2个月后降至50%,1岁时不超过5%,2岁时即降至正常成人水平(0~2%),珠蛋白生成障碍性贫血时,由于遗传缺陷,珠蛋白基因发生突变,致珠蛋白肽链合成障碍,根据合成受抑制的珠蛋白肽链的不同,可分为若干类型,最常见的是β链合成障碍,称β-珠蛋白生成障碍性贫血,其次为α链合成减少的α珠蛋白生成障碍性贫血,还有较少见的δ链合成减少或δ与β链皆减少的δ或δβ以及γβ珠蛋白生成障碍性贫血,珠蛋白生成障碍性贫血的分类有两种方法:
①根据珠蛋白肽链基因缺陷情况分为α,β,γ和δ等,临床上以α,β两类最为常见。
②根据临床病情的轻重程度分为静止型,轻型,中间型及重型,目前多采用两种分类法联合分类。
1.α珠蛋白生成障碍性贫血
编码α珠蛋白的基因位于第16号染色体短臂末端(16p13.3),每条染色体上有2个α基因α1和α2,故二倍体细胞中共有4个α基因,根据α基因缺失的数量和功能障碍的情况不同又可分为5种类型:
(1)静止型珠蛋白生成障碍性贫血:又称α2-珠蛋白生成障碍性贫血或α -珠蛋白生成障碍性贫血,由一条染色体上缺乏一个α基因(-,α/α,α);其临床特点为患者无症状,红细胞形态正常,出生时血中Hb Bart′s(γ4)1%~2%,3个月后即消失,此型诊断困难。
(2)α1-珠蛋白生成障碍性贫血或α0-珠蛋白生成障碍性贫血:本病也称标准型α-珠蛋白生成障碍性贫血,由一条染色体上的两个α基因缺失(-,-/α,α)或2个α 珠蛋白生成障碍性贫血基因(-,α/-,α)导致α链合成障碍,其临床特点为患者无症状,红细胞形态有轻度改变;HbF正常,出生时,HbBart占5%~6%,3个月后即消失;此型多见于HbBart胎儿水肿综合征患者的双亲和HbH病的双亲中一人,本病不需治疗。
(3)血红蛋白H病:是α-珠蛋白生成障碍性贫血的中间型,由一条16号染色体上的一对α基因和另一条16号染色体上的一个α基因缺陷,基因型为(-,α/-,-),临床特点为出现中等度或严重小细胞低色素性贫血,包涵体试验阳性,新生儿期血中HbBart 20%~30%,婴儿期以后才出现症状者,出现不同程度贫血,黄疸,肝脾肿大,发作性加重(尤以感染和药物为诱因);成熟红细胞形态改变明显;年长儿童则可出现Hb H(β4)4%~20%,而HbA2及HbF含量正常,
(4)HbBart胎儿水肿综合征:是重型α-珠蛋白生成障碍性贫血,为α1的纯合子状态,所有2条16号染色体上的α基因均缺陷,其基因型为(-,-/-,-),由于控制α链合成的4个基因均缺失,故无α链合成,因此不能合成含α链的HbA,HbA2和HbF,在胎儿后期γ,β链各自形成大量的γ4(HbBart)和β4(HbH),同时胚胎早期ζ链合成代偿性增加并持续至整个胎儿期,并与α组成HbPortland,HbBart具高度氧亲和力且极不稳定,导致宫内胎儿严重的慢性溶血和组织严重缺氧,心力衰竭,水肿,造成流产,死胎,绝大多数于妊娠期30~40周时胎儿死于宫内或娩出后短期内死亡,一旦胎儿有幸出生时全身重度水肿,腹水,重度贫血,轻度黄疸,肝大;HbBart含量70%~100%,可同时少量HbH(β4),本症双亲为α1珠蛋白生成障碍性贫血杂合子,同胞中的发病率约为1/4。
(5)非缺失型α-珠蛋白生成障碍性贫血:α基因结构并未缺失,但其功能障碍,表达水平降低,临床出现与α-珠蛋白生成障碍性贫血类似的表现,其基因型为(a,aThal/a,aA)或非缺失型双重杂合子(a,aThal/a,aThal)。
2.β珠蛋白生成障碍性贫血
β珠蛋白基因位于第11号染色体短臂1区2带(11p1.2),本病除少数几种为几个核苷酸缺失外,绝大部分由点突变所致,目前已发现β基因突变有数十种,β链合成部分受抑制者称为β 珠蛋白生成障碍性贫血,β链合成完全受阻者称为β0珠蛋白生成障碍性贫血,肽链合成抑制涉及δ链者称为δβ珠蛋白生成障碍性贫血δβ 或δβ0,染色体上的2个等位基因突变点相同者称为纯合子;同源染色体上只有1个突变点者称为杂合子;等位基因的突变点不同者称为双重杂合子,我国β-珠蛋白生成障碍性贫血的发生率为0.67%,以广东,广西,云南,贵州等省地为高。
(二)发病机制
1.地中海贫血
HbBart胎儿水肿综合征缺失4个α肽链基因,完全没有α肽链生成,在胎儿期合成的γ链可聚合成Hb Bart(γ4),HbBart的氧亲和力高,造成组织严重缺氧和水肿,HbH病有3个α肽链基因缺失或缺陷,仅有少量α肽链生成,在胎儿期,肽链与γ肽链结合成少量Hb F(α2γ2),故能存活至出生,出生后随年龄增长,γ肽链的合成转为β肽链的合成,未能与α肽链结合的β肽链聚合成HbH(β4),HbH的氧亲和力高,失去运氧功能,游离的β肽链在红细胞内易变性形成Hb H包涵体,使红细胞膜受损害,标准型α地中海贫血和静止型α地中海贫血,分别缺失2个和1个α肽链基因,肽链生成稍微减少。
2.β地中海贫血
β地中海贫血是β肽链合成障碍,但α肽链合成正常,以致Hb A(α2β2)生成减少,尽管相对增多的α肽链可与代偿产生较多的δ肽链和γ肽链结合,可使Hb A2(α2δ2)和Hb F(α2γ2)增加,但仍有大量多余游离的α肽链沉淀于红细胞内,形成包涵体,使红细胞膜受损致幼红细胞在骨髓内过早破坏,造成“无效红细胞生成”;流入血循环的幼红细胞过早在脾和肝内受破坏,造成溶血性贫血,继而骨髓和骨髓外造血功能亢进,骨髓增生活跃,肝脾肿大,长期骨髓增生,引起骨髓腔扩大,使骨骼生长畸形和发育障碍,另外,严重的溶血性贫血,需反复输血,可有血红素和铁的积蓄过多,并发含铁血黄素沉着症。
诊断
根据婴儿期开始呈慢性进行性贫血,脾脏肿大和特殊面容,外周血红细胞形态改变,红细胞渗透脆性减低和(或)阳性家庭史,应高度怀疑本病,进一步作血红蛋白电泳,基因分析等检查,可以确诊。
鉴别诊断
须与缺铁性贫血,遗传性球形细胞增多症,病毒性肝炎或肝硬化,G-6-PD缺陷症,新生儿同族免疫性溶血病鉴别。
小儿珠蛋白生成障碍性贫血西医治疗
(一)治疗
1.一般治疗
适当注意休息和营养,预防感染可防止贫血加剧,轻型病例无明显症状者仅给予一般治疗即可。
2.对症治疗
(1)输血:是重型β-珠蛋白生成障碍性贫血有效疗法。国外采用高量输血,开始每周输1次,可输新鲜或经等渗盐水洗涤过的红细胞或压缩红细胞,反复多次使Hb升至120~140g/L。然后定期每隔4~5周1次,每次输入20ml/kg或压缩红细胞15ml/kg,维持Hb>80g/L,这样使病人保持较高的Hb水平,以消除贫血症状,防止严重并发症发生。
(2)铁螯合剂:反复输血可导致继发性含铁血黄素沉着症,如反复输血>100次,血清铁饱和度>80%者应给予螯合剂去铁胺(deferoxamine),去铁胺可与体内过量三价铁形成,在注射用水或生理盐水中静滴,可于晚上睡眠时给予,每次维持8h,每周连用5~6晚;也可采用肌注或便携式输液泵作腹壁皮下注射,8~12h给予,主要不良反应为皮疹,药物过量可致长骨生长障碍,白内障等,口服螯合剂deferiprone也可应用,可促进铁的排出,推荐剂量为每天75~100mg/kg,主要不良反应为粒细胞减少,关节炎,肌肉痛等,少部分病例出现胃肠道反应,严重者应停药,铁螯合剂应用时,宜用大剂量维生素C口服以增加尿铁的排出。
(3)抗氧化剂治疗:可长期口服维生素E 50~150mg/d,有抗红细胞膜脂质的过氧化损伤,减少溶血作用。
(4)脾切除或脾栓塞术:可改善贫血症状和减少输血次数,适用于:
①输血日益增多者;
②巨脾引起压迫症状者;
③合并脾功能亢进者,脾切除一般于5~6岁后施行,本疗法可减轻贫血症状和减少输血量。
3.基因活化疗法
应用化学药物重新活化γ珠蛋白的基因,与过量α珠蛋白结合使HbF的合成增加,改善β-珠蛋白生成障碍性贫血的症状,化学药物有羟基脲每天15mg/kg,每3周为1个疗程,可重复使用;有报道连续使用3年而无明显毒性不良反应,此外可用阿糖胞苷,白消安(马利兰),长春新碱等,也有应用异烟肼每天15~20mg/kg,每3周为1个疗程,可使过量的α珠蛋白减少。
4.造血干细胞移植
异基因骨髓或脐血干细胞移植成功后的5年无病生存率达80%以上,可采用HLA匹配的同胞姊妹,父母或者非亲缘供者的骨髓,G-CSF,动员外周血以及脐血造血干细胞。
5.基因治疗
目前正在研究中。
(二)预后
β-地中海贫血重型并发症常是导致患儿死亡的重要原因,如不治疗,多于5岁前死亡,中间型和轻型地中海贫血正确处理可长期存活,α-地中海贫血,HbBart胎儿水肿综合征,常发生早产,死产或生后不久死亡,静止型,标准型预后好,HbH病正确处理亦预后较好。
小儿珠蛋白生成障碍性贫血中医治疗
当前疾病暂无相关疗法。
高蛋白低脂肪
对一般贫血病人来说 ,首先应考虑给予高蛋白饮食.这可以通过食用动物的瘦肉以及肝,肾等内脏 ,获得优质蛋白的补充.其次 ,应尽量控制脂肪的摄入量.因为脂肪可抑制人体的造血功能 ,高脂肪还可导致腹泻,消化不良,肥胖病等疾患。
丰富的维生素
饮食结构中维生素的含量丰富 ,对各类疾病的患者都是适宜的.就贫血病人而言 ,维生素 B 1,维生素 B 12,维生素 C和叶酸等是至关重要的.维生素 B 1的补充 ,可以通过粮食特别是粗杂粮食物获得;维生素 B 12和叶酸 ,主要来源于动物内脏等食物;维生素 C的主要来源 ,则是各种新鲜的蔬菜和水果。
补充微量元素
食用含铁丰富的食物 ,几乎成为贫血病人的常识.值得注意的是 ,适量补充微量元素铜对纠正贫血也相当重要 ,不过人体对锏的生理需要量甚微 ,通过日常饮食即可满足。但是,如果饮食营养欠佳 ,而又少食甚至不食蔬菜 ,就会给纠正贫血带来不利。
少食含盐食物
贫血病人应少食含盐食物为好 ,一旦出现水肿还应暂时禁盐。
本病为遗传性疾病,开展人群普查,遗传咨询,做好婚前指导工作,避免患者之间联姻对预防本病均有重要意义,近年来采用新生儿脐血做pH 8.34不连续醋纤膜微量电泳,可作为一种早期筛查α珠蛋白生成障碍性贫血的方法,尤其是可检出无临床症状的静止型和标准型α珠蛋白生成障碍性贫血,开展产前诊断,用胎儿血测定α/β链比例或从胎血白细胞或绒毛细胞提取DNA制作基因图可及早确诊本病,及时终止妊娠,可减轻家庭和社会的负担,提高民族素质。
1.血象
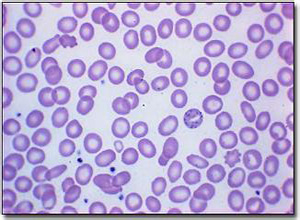
由于Hb合成下降而呈小细胞低色素性贫血,外周血片可见红细胞大小不等,中央浅染区扩大,红细胞形态改变明显,出现异形(梨形,泪滴状,小球形,三角形或靶形),碎片红细胞和有核红细胞,嗜碱性点彩现象等,嗜多染性红细胞,豪-周小体等,网织红细胞正常或增多,通常≤10%。
2.红细胞渗透脆性试验
减低,0.40%~0.38%NaCl溶液开始溶血,在0.20%或更低的低渗盐水中才完全溶血,轻型病例可正常。
3.HbF测定
这是诊断重型β珠蛋白生成障碍性贫血的重要依据,HbF含量轻度升高(<5%)或明显增高(20%~99.6%);HbA2常降低、正常或中度增高,HbA2 3.5%~8.0%。
4.血红蛋白电泳
分离出HbH或Hb Bart是确诊α蛋白生成障碍性贫血的重要依据。
5.肽链分析
采用高效液相层析分析法可测定α,β,γ,δ肽链的含量,Cooley贫血时,β/α比值<0.1(正常值为1.0~1.1)。
6.异丙醇试验
呈阳性,如同热不稳定试验一样,可鉴别不稳定Hb和α-海洋性贫血。
7.包涵体生成试验
红细胞包涵体和Heinz小体可呈阳性。
8.骨髓象
红系增生明显活跃,以中,晚幼红细胞占多数,成熟红细胞形态改变与外周血相同,但α-珠蛋白生成障碍性贫血静止型骨髓象可正常。
9.核酸分析
测定Hb肽键的mRNA含量或通过DNA分子杂交及限制性内切酶技术鉴定患者的珠蛋白基因是否缺失,近年来应用限制性片段长度多态性(RFLP)连续分析,人工合成的寡核苷酸探针杂交及基因体外扩增(PCR)技术间接或直接进行基因诊断,可检测和鉴定突变基因, 常规做X线,B超,心电图等检查,骨骼X线检查,骨髓腔增宽,皮质变薄和骨质疏松,颅骨的内外板变薄,颅骨骨髓腔增大,板障加宽,骨皮质间髓梁有垂直条纹,呈短发状改变,短骨由于骨小梁变薄而成花边或嵌花样间隔,以指骨及掌骨出现较早,长骨此质变薄而髓腔变宽,以股骨无端较明显。
可并发黄疸,肝脾肿大,胆石,可并发溶血危象,水肿,腹水,贫血,骨骼改变,生长发育停滞,常并发支气管或肺炎,并发含铁血黄素沉着症,造成脏器损害,并发心力衰竭,肝纤维化,肝功衰竭等。