
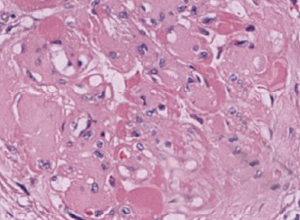
肾小球病例形态改变,IGA肾病的表现之一。IgA肾病又称Berger病,是一种特殊类型的肾小球肾炎,多发于儿童和青年,发病前常有上呼吸道感染,病变特点是肾小球系膜增生,用免疫荧光法检查可见系膜区有IgA沉积。
病理变化:病变程度差异很大,早期病变轻微,呈局灶性,仅少数肾小球有轻度系膜增宽和阶段性增生,局灶性增生性改变可发展为局灶性硬化。有些病变较明显,可有弥漫性系膜增生,偶尔可有新月体形成。最突出的特点是免疫荧光显示系膜区有lgA沉积,并同时并有 C3,lgG和IgM较少,电镜观察证实系膜区有电子致密物沉积。
临床病理联系:主要症状为镜下或肉眼复发性血尿,可伴有轻度蛋白尿。少数病人出现肾病综合征。
IgA肾病多呈慢性进行性过程,约半数病人病变逐渐发展,可出现慢性肾功能不全。
IgA肾病是一组多病因引起的具有相同免疫病理学特征的慢性肾小球疾病。临床上约40%—45%的患者表现为肉眼或显微镜下血尿,35%—40%的患者表现为显微镜下血尿伴蛋白尿,其余表现为肾病综合征和肾功能衰竭。IgA肾病肾病是世界范围内一种常见的肾小球疾病,IgA肾病的流行在不同洲、不同国家或在一个国家不同地区的差异很大,如亚洲的日本、新加坡,IgA肾病肾病的发病率占原发性肾小球疾病的50%,而美国西部的印第安人低发区只占2%。一般而言,白人、黄种人明显高于黑人的发病率。我国IgA肾病的发病率占原发性肾小球疾病的26% ̄34%。男女之比大约是2:1。 以血尿为主的IgA肾病目前尚无特效的治疗。由于IgA肾病的病理类型及肾小球受损程度的差异较大。因此,应严密观察患者肉眼血尿发作的频率、蛋白尿的程度、有无高血压及肾功能受损程度,而分别采取相应的防治措施。
从上面可以看出:IgA肾病不是恶性的。
与IGA肾病预后有关的因素:
(1)男性患者,起病年龄较大者预后差。
(2)持续性镜下血尿伴有蛋白尿,预后差。
(3)中、重度蛋白尿常提示最终发展到肾功能不全,预后较差。但IgA肾病表现为肾病综合征的患者,若肾组织病理变化轻微,对糖皮质激素治疗反应好,预后好。
(4)IgA肾病患者有高血压,特别是难于控制的严重高血压,预后差。
(5)妊娠对IgA肾病患者的影响,无高血压及肾功能减退的IgA肾病患者,妊娠一般是安全的。
1.反复发作性肉眼血尿或持续性镜下血尿;不伴水肿、高血压或其他肾功能异常。
2.血中IgA 水平升高;肾组织免疫荧光检测有显著IgA 沉着于系膜区。
3.除外能引起系膜IgA沉积的过敏性紫癜等其他疾患:
a.发作性肉眼血尿:多见于儿童。其肉眼血尿多在上呼吸道感染(扁桃体炎等)后发生,亦有部分在急性胃肠炎或尿路感染后发作,间隔时间多在24~72小时。肉眼血尿可持续数小时至数天,然后转为持续性镜下血尿,部分病人血尿可消失,但常发作,发作时重现肉眼血尿,可伴有轻微全身症状,如肌肉酸痛、尿痛、腰骨痛,或一过性血压及尿素氮升高。
b.镜下血尿及无症状性蛋白尿:此为儿童及青少年IgA肾病的主要临床表现,常在体检中被发现,可表现为单纯镜下血尿,或镜下血尿伴少量蛋白尿。
c.蛋白尿:为轻度蛋白尿,尿蛋白定量一般〈1g/24h,少数患者可出现大量蛋白尿甚至出现肾病综合征。
d.其他:部分IgA肾病患者可出现肾病综合征,急进性肾炎综合征,肾功能衰竭,少数可出现腰和/或腹部剧痛伴血尿。
肾小球体积增大:肾小球体积增大是指肾小球由于脂蛋白肾小球病引起的肾小球的体积增大的生理病理上的特征性的形态学改变。脂蛋白肾小球病(lipoproteinglomerulopathy)是一种肾脏疾病,其病理特征为肾小球毛细血管襻腔中存在脂蛋白栓子,肾外无脂蛋白栓塞表现。脂蛋白肾小球病多见于男性,男女比例为15∶8;平均发病年龄为32岁(4~49岁)。多数病例为散发性,少数为家族性发病。脂蛋白肾小球病(lipoproteinglomerulopathy)是一种肾脏疾病,其病理特征为肾小球毛细血管襻腔中存在脂蛋白栓子,肾外无脂蛋白栓塞表现。临床表现类似于Ⅲ型高脂血症,伴以血浆载脂蛋白E(apoE)升高。本病是1987年SaitoT等人在第17届日本肾脏病学会地区年会上首次报道,1989年Sakaguchi等人根据病人的临床表现、病理学特点,提出将本病作为一个独立的肾小球疾病,并于同一年将本病命名为脂蛋白肾小球病。然而,脂蛋白肾小球病通过降血脂治疗并不能改善肾小球病变。目前认为本病是原发于肾小球Ⅲ型仅限于肾脏有脂质沉着的罕见病。
肾小球基底膜蛾噬现象:肾小球基底膜蛾噬现象是指甲-髌骨综合症的诊断依据之一。对肾活检标本进行判断不能仅用肾小球基底膜蛾噬现象,必须用磷钨酸染色鉴定原纤维因其敏感性更高,对诊断更有价值。指甲-髌骨综合征为遗传性疾病其传递方式是常染色体显性遗传。发生率为4.5/100万~22/100万,没有性别的差异。患者传递给其后代的机会为50%,基因位点与腺苷酸环化酶和ABO血型的位点连锁位于9号染色体上。Looij等根据自己的数据和资料推断,如果本综合征家族的一个患者有明显的临床肾脏表现,则其所生孩子患肾病的危险性是1/4,发展至肾功能衰竭的概率是1/10。
肾小球旁器增生:肾小球旁器增生综合征,别名:巴特综合征。巴特尔综合征;Bartter综合征;先天性醛固酮增多症;慢性特发性低钾血症;Barttersyndrome。本综合征是一常染色体隐性遗传病,由Bartter(1962)首次报道故称为Bartter综合征。肾小球旁器增生综合征以严重的低血钾、碱中毒为主血钠、氯均低,血压正常伴多饮多尿、便秘、脱水。血浆肾素-血管紧张素及醛固酮均升高。本综合征是一常染色体隐性遗传病,由Bartter(1962)首次报道故称为Bartter综合征。其临床特征为严重的低钾血症和代谢性碱中毒,伴有高肾素高醛固酮血症、肾小球旁器增生和肥大及肾小管保钠和浓缩功能障碍,但无高血压及水肿且对外源性血管紧张素Ⅱ无反应。认为本综合征是由离子通道基因突变引起的临床综合征。本病又称为先天性醛固酮增多症慢性特发性低钾血症肾小球旁器增生综合征近年来分子诊断学研究揭示Bartter综合征有3种不同的临床和遗传类型,即先天性Bartter综合征典型Bartter综合征和Gitelman综合征。通常所说的Bartter综合征是指典型Bartter综合征。先天性Bartter综合征病人发现有两种基因型,Ⅰ型是由于N+-K+-2CL-发生失功能性基因突变所致Ⅱ型是由于ROMK基因突变所致典型Bartter综合征是由于CLC-kb通道基因突变所致。
肾小球硬化:局灶性肾小球硬化症(focal glomeruloscerosis)是指肾小球毛细血管袢有局灶性节段性硬化或透明变性,无明显细胞增生的一类肾小球毛细血管。可作为系膜增生、系膜IgM沉积和局灶性肾小球硬化,可是微小病变性肾病对类固醇耐药,反复发作慢性进展的后果。亦有对激素无效的原发性肾病综合征早期肾活检即为局灶性肾小球硬化。故对本病是否作为一种独立的肾小球疾病尚有争论。但从代表一种与其他肾脏病不同的临床病理类型,亦可作为一独立的疾病,较为常见,且有逐渐增加的趋势。
1、劳逸结合:因劳累过度,剧烈运动,常可使血尿增加,故应做到起居有节,注意卧床休息,适度锻炼身体,防止熬夜、过度疲劳及剧烈运动。
2、防治炎性疾病:积极消除易感和诱发因素,如上呼吸道、皮肤、肠道、尿路感染,根治疮疖,真菌感染,对反复因扁桃体炎而诱发血尿发作者,可行扁桃体切除术,儿童包皮过长者宜适时环切。一旦出现炎症感染,积极治疗。
3、精神调养:凡患尿血的病人,均有不同程度的精神紧张、抑郁和悲观。因此,在日常生活中,要时时注意言行,慌张、高叫等都会增加病人的不安和恐惧心理,故精神调养显得尤为重要。尽可能减少对病人不良的精神刺激,保持心情舒畅,以利于疾病的康复。
4、预防外感:本病常因上呼吸道感染、扁桃体炎而使病情加重,故应预防感冒,如体质较差,容易感冒者,可适度锻炼身体,增加抵抗力,防止上呼吸道感染发生,并服用中药玉屏风散以益气固表。
5、预防肾功能不全:影响IgA肾病长期预后的因素很多,最常见于高龄男性起病者,或持续性血尿伴有大量蛋白尿者,或伴有严重高血压患者等,对此类情况,应严密观察,高度重视,并给予及时合理的防护措施,由此才可阻滞IgA肾病发展至肾功能衰竭的进程,达到治病防变的目的。