
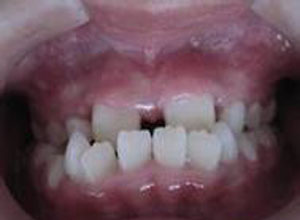
门齿间距增宽是甘露糖苷贮积症Ⅱ型的症状之一。甘露糖苷贮积症Ⅱ型多于2 岁后发病,体格与精神运动发育正常,2 岁后开始进行性大脑发育迟缓,频繁的呼吸道感染,面容丑陋,眉粗厚,门齿间距增宽,凸颌,前发际低,有轻度双侧耳聋(多为感觉性)。部分病人可有全血减少。
遗传基因缺陷,病人体内酸性型α-甘露糖苷酶缺陷是主要病因。是因α-甘露糖苷酶缺乏所引起的全身性疾病临床特征类似Hurler综合征,无黏多糖尿但组织中含甘露糖的成分增加。
正常时这种酶水解低聚糖(oligosaccharide)α-键连接的甘露糖苷。因为该病酸性型α-甘露糖苷酶缺陷,故糖蛋白不能分解,富有甘露糖的低聚糖即沉积于组织内,主要在脑内,且随尿排出。脑内神经元肿胀,呈气球样,其中的沉积物质就是含甘露糖的糖蛋白。
根据临床症状,X线表现,反复感染,智力低下,运动迟钝,肝及其他组织活检显示酸性型α-甘露糖苷酶缺乏和尿中无过多黏多糖排出等,即可诊断。
1、周围血象中的中性粒细胞、淋巴细胞及骨髓细胞内可见有空泡肝组织,和其他组织活检,生化分析,显示甘露糖苷增加,及酸性型α-甘露糖苷酶缺乏;尿中含甘露糖的寡糖。
2、X线检查:X线表现为轻度多发性骨发育不全,腰椎椎体发育不全,呈鸟嘴状;髂骨翼轻度外翻,髋外翻畸形,肋骨增宽,长骨的骨骺骨干,掌骨和指骨增粗;颅骨穹窿部及颅底硬化,有的病例可出现严重脊柱侧弯和驼背。
应与下面的情况相鉴别:
Ⅰ型或婴儿型出生时多发育正常,1岁左右可出现进行性面容丑陋,巨舌,扁鼻,大耳,牙缝宽,头大,手足大,四肢肌张力低下并运动迟钝,但其程度不及Hurler综合征,胸骨隆凸,胸腰驼背,颅盖骨增厚,角膜一般清晰,但也有的患者出现晶体浑浊,初生时部分患者就有耳聋或有语言障碍,智力低下。
根据临床症状,X线表现,反复感染,智力低下,运动迟钝,肝及其他组织活检显示酸性型α-甘露糖苷酶缺乏和尿中无过多黏多糖排出等,即可诊断。
人可有全血减少。
预防:
1、一级预防
(1)婚前检查。
(2)遗传咨询。
2、遗传病治疗中总的原则是禁其所忌,去其所余,补其所缺,调节代谢平衡,防止症状的出现。
(1)纠正代谢紊乱:这是目前治疗遗传性代谢病的最主要方法,随着对遗传性代谢病发病机制和中间过程的认识不断深化,此法的适用范围也日益扩大。
(2)纠正酶活性异常。