(一)发病原因
本症为遗传性疾病,多属性连锁隐性遗传,个别为染色隐性遗传。
(二)发病机制
应用分子生物学方法已将DMD的基因定位于X染色体Xp21.1~Xp21.3,致病基因为dystrophin基因,它是至今发现的最大的人类基因,约2000kb以上,含有79个外显子编码,1个14kb的转录区。研究表明60%~70%的DMD是由于基因缺失或重复突变所致。
基因缺失呈非随机性分布,主要发生在基因的中央区(80%),少数发生在5端(20%)。大的基因缺失常常开始于基因的5端,基因缺失造成开放的读码框的破坏,导致DMD表现。BMD患者,缺失基因保持了翻译读码框,并能产生一个具有一半功能、长度缩短的蛋白质。“读码框”假说解释了92%的DMD/BMD患者不同的临床类型。
dystrophin是dystrophin糖蛋白复合物(DGC)的一部分,DGC是膜相关蛋白的综合体,跨越肌纤维膜,连接细胞内的细胞骨架和细胞外的基质。duchenne肌营养不良患者,由于dystrophin的缺失导致DGC成分的减少,虽能正常合成但不能正确的装配和整合至肌纤维膜上。由此推测由于DGC的受损,引发一系列连锁反应,导致DMD的肌细胞坏死。dystrophin的缺乏使肌纤维膜下的细胞骨架和细胞外基质的联系受到破坏,造成肌纤维膜不稳定,膜撕裂,肌细胞坏死。
常在10岁以后起病,首发症状为骨盆带及股部肌肉力弱,进展缓慢,病程长,出现症状后25年或25年以上才不能行走,多数在30-40岁时仍不发生瘫痪,预后较好。
1.血清生化检查
肌酸磷酸激酶(CK)明显升高,达1.5万~2万U/L,甚至更高。血清CK升高可出现于出生时,疾病后期略有降低。
2.肌肉活体组织检查
特征性的病理改变有散在的退行性变和坏死肌纤维。随着时间的延长,出现肌内膜结缔组织增加以及肌纤维的丧失,脂肪组织的替代。
3.基因诊断
DMD基因定位于Xp21.1-21.3,基因编码的蛋白质为dystrophin。20世纪90年代后国内各大医院应用核素DNA印迹法杂交、缺失热点外显子的聚合酶链反应(PCR)分析进行基因诊断,缺失检出率为56.7%~63.0%,应用DMD基因缺失热点9对引物PCR分析,缺失型大宗病例的检出率为47.5%~49.6%。国内已应用定量PCR测定、短串联重复序列连锁分析检出DMD基因携带者。对于点突变型DMD的诊断尚缺乏系统的研究。
4.肌电图
为肌源性改变,病变肌肉呈低电位,波形持续时间缩短,而多相波增高。其他尚应做心电图、脑电图等检查。
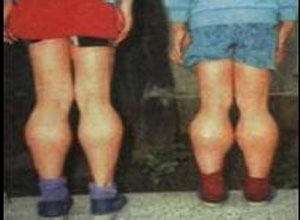
Duchenne型营养不良症(DMD):也称严重性假肥大型营养不良症,几乎仅见于男孩,母亲若为基因携带者,50%男性子代发病,常起病于2-8岁,初期感走路苯拙,易于跌倒,不能奔跑及登楼,站立时脊髓前凸,腹部挺出,两足撇开,步行缓慢摇摆,呈特殊的“鸭步”步态,当由仰卧走立时非常困难,必先翻身俯卧,再双手攀缘两膝,逐渐向上支撑起立(Gower征)。亦可见于肢近端肌肉、股四头肌及臂肌。
常在10岁以后起病,首发症状为骨盆带及股部肌肉力弱,进展缓慢,病程长,出现症状后25年或25年以上才不能行走,多数在30-40岁时仍不发生瘫痪,预后较好。
由于本病的致病基因位于Xp21.1,致抗肌营养不良蛋白(dystrophin)缺乏,目前尚未找到一种可逆转本病病程的特效疗法。故应强调采取有效的措施降低本病在人群中的遗传负荷,减少本病的发生。
1.检出携带者
极早发现携带者,指导其婚姻和生育,可减少进行性肌营养不良患儿和携带者的出生。检出携带者的方法有多种,如家系调查、血清酶活性测定、肌电图检查和肌活检等。其中以测定血清CPK及CPK-MB活性最为简便、实用。上述几种方法综合应用则可提高检出携带者的阳性率。
2.产前诊断
有性连锁隐性遗传史或携带者或已生一患儿的女性,在妊娠时,如为男婴,受累者约占半数,此时可抽取胎盘血清做CPK测定,如升高明显,或PCR检测病理基因阳性,应做人工流产。阴性或为女婴,则可让其出生。
3.新生儿筛选
目的是及早发现患儿及携带者。以生后6~10周后测定CPK活性为首选。