肝豆状核变性常见病
肝豆状核变性(hepatolenticular degeneration)又称Wilson病,本病于1911年首先由Wilson报道,此为一种常染色体隐性遗传性疾病,青少年多见,是先天性铜代谢障碍性疾病。由Wilson首先报道和描述,是一种遗传性铜代谢障碍所致的肝硬化和以基底节为主的脑部变性疾病。临床上以肝损害、锥体外系症状与角膜色素环等为主要表现。早期(尤其是症状前)诊断和及时、确切的治疗常可获得与健康人一样的生活和寿命。
肝豆状核变性(hepatolenticular degeneration)又称Wilson病,本病于1911年首先由Wilson报道,此为一种常染色体隐性遗传性疾病,青少年多见,是先天性铜代谢障碍性疾病。由Wilson首先报道和描述,是一种遗传性铜代谢障碍所致的肝硬化和以基底节为主的脑部变性疾病。临床上以肝损害、锥体外系症状与角膜色素环等为主要表现。早期(尤其是症状前)诊断和及时、确切的治疗常可获得与健康人一样的生活和寿命。
(一)发病原因
肝豆状核变性系常染色体隐性遗传性疾病,受累基因与铜代谢紊乱有关,与位于染色体的酯酶D基因与视网膜母细胞瘤基因紧密连锁。
(二)发病机制
WD的发病机制有胆道排泄减少、铜蓝蛋白合成障碍、溶酶体缺陷、金属巯蛋白基因异常及调节基因异常等学说,目前以前二种学说获得多数学者赞同。
1、铜代谢合成障碍
多数实验室用64Cu对体内铜代谢研究证明,血清铜蓝蛋白减少是WD体内铜积蓄的主要原因。但铜蓝蛋白为何缺乏,尚未完全阐明。Bichtrrich根据铜蓝蛋白电泳发现,正常成人是由先构成的未分化的铜蓝蛋白D在肝脏内经肽酶将其大部分转化为铜蓝蛋白C,然后由80%铜蓝蛋白C与20%铜蓝蛋白D构成铜蓝蛋白,而WD患者仅存在铜蓝蛋白D,而几乎没有C部分,故引致铜蓝蛋白合成障碍。
2、胆道铜排泄障碍
正常成人每日需从食物中吸收铜2~5mg,铜离子进人体内后,大部分先与白蛋白疏松结合为直接反应铜,运送到肝脏,在肝细胞内转与各种球蛋白,主要是α2-球蛋白牢固地结合为铜蓝蛋白(间接反应铜)。一般血浆中的总铜量90%~95%以铜蓝蛋白形式存在,仅约5%的铜与白蛋白、氨基酸和多肽疏松结合存在,后者除在各脏器内自由通过细胞膜与血浆铜交换外,大部分由溶酶体摄取经胆管从粪便中排出,少数由尿排出。即正常人从食物中吸收的铜,除体内生理需要外,过剩的铜绝大部分从胆管中胆汁排泄。Frommer率先测定8例WD患者及10例对照组的十二指肠液内含铜量,发现WD组显著低于对照组,提出胆管排铜障碍是造成WD患者体内铜蓄积的重要原因。
铜是人体必需的微量元素,作为辅基参与多种重要生物酶合成。正常成人每日从饮食摄取铜2~5mg,约30%在胃、十二指肠及空肠上端吸收入血,大部分与白蛋白疏松结合进入肝细胞,在肝细胞中铜与α2球蛋白牢固结合成铜蓝蛋白(ceruloplasmin,CP),CP有氧化酶活性,呈深蓝色,剩余铜被结合到其他特殊铜蛋白中。正常人每日胆汁排铜量约1200μg。约70%的CP存在于血浆,其余存在于血管外,血液循环中90%~95%的铜结合在CP上。CP有重要生理功能,可作为铜的供体参与细胞色素C及其他铜蛋白合成,具有亚铁氧化酶作用,将亚铁氧化为高铁状态,使氧还原成水。剩余的铜通过胆汁、尿液和汗液排出体外。WD患者铜蓝蛋白合成障碍,90%以上患者血清CP量明显减少,但肝内前铜蓝蛋白(Apo-CP)含量及结构正常,提示生化障碍发生在肝内Apo-CP与铜结合环节,CP合成障碍是本病基本的遗传缺陷。肝内铜代谢紊乱引起血清CP合成障碍,导致血清铜及CP降低,尿铜排泄增多,胆道排铜减少,过量铜在肝脏、脑、肾脏及角膜等组织沉积致病,但约5%的Wilson病人血清CP水平正常难以解释。
近年研究已确定CP基因位于13号染色体(13q14-21),有多种突变型,表达CP是132kD糖蛋白,由1046个氨基酸残基组成单条多肽链,结合6个三种不同类型铜离子。WD患者铜蓝蛋白前体无异常,基因及表达产物无变化,从遗传基因角度不能解释WD血清CP明显减少。基因突变有明显遗传异质性,突变方式包括转换(A→G)、颠换(C→G)、缺失(CCC→CC)及插入(T→TT),其中C→G颠换最常见,造成编码氨基酸变化(如组氨酸变成谷氨酸、天门冬氨酸变成丝氨酸)及移码突变,至今发现的突变均涉及ATP酶功能区。WD基因突变引起编码P型ATP酶(也称ATP7B)功能改变,ATP7B主要功能是铜转运,部分或全部功能丧失,不能将多余铜离子从细胞内转运出去,使铜离子在特定器官和组织沉积致病。
WD的分子发病机制存在种族差异,欧美患者ATP7B基因高频突变点是14号外显子,处于ATP7B基因磷酸化区及ATP结合区,两个功能区基因突变使功能消失,导致酶缺乏,转运过程中能量引起铜离子在细胞内滞留。中国WD患者高频突变点8号外显子在整个ATP7B基因中处于跨膜功能区,引起蛋白质一级、二级结构改变,导致细胞膜铜转运停滞而致病。
WD的病理表现为大量的铜沉积于组织。病变特征性地分布于脑组织、肝脏、肾脏及角膜等处。脑病变以壳核最早和明显,其次为苍白球、尾状核及大脑皮质,丘脑底核、红核、黑质、丘脑及齿状核亦可受累。神经元显著减少或完全脱失,轴突变性和星形胶质细胞增生。角膜边缘后弹力层及内皮细胞浆内,可见棕黄色细小铜颗粒沉积,严重者角膜中央区及间质细胞中也可见到。肝脏外表及切面可见大小不等结节或假小叶,颇似坏死后肝硬化,肝细胞脂肪变性,含铜颗粒。电镜下可见肝细胞内线粒体致密、线粒体嵴消失及粗面内质网断裂等。
1、铜含量测定
(1)头发铜含量测定 对肝豆状核变性诊断及鉴别诊断的价值不大。
(2)肌肉合铜量测定 部分诊断困难的拟诊肝豆状核变性患者有一定的参考价值。
(3)指甲含铜量测定 指甲含铜量测定是一种无损伤性的检查方法,其优缺点与头发铜测定相同。
(4)胆汁内含铜量测定 对肝豆状核变性的诊断有特异价值。肝豆状核变性患者胆汁内含铜量显著减低。
2、影像学检查
(1)肝豆状核变性的肝脏B超检查 有其特殊的声像图,并将肝实质的声像图按肝损害的不同程度依次分为光点闪烁型、岩层征型、树枝状光带型和结节型,对肝豆状核变性具有特征性诊断价值。对尚未出现神经症状的肝豆状核变性肝硬化者(结节型)与慢性肝炎肝硬化者有鉴别价值。可评估脾脏大小、形态。可显示胆结石、肾结石、肾钙质沉着。
(2)食管钡剂造影摄片 脾门静脉造影或动脉造影可对疑有门脉高压临床表现的肝豆状核变性患者进一步确诊,有助于治疗方案的制订。
(3)骨关节X线检查 在肝豆状核变性诊断上的意义:①骨关节X线改变是本病潜在的诊断指标。临床上难以确诊病例,不管有无骨关节症状,都可利用该检查帮助诊断。②在儿童、少年期出现不明原因的病理性骨折或X线照片发现腕、膝关节异常,要考虑到患肝豆状核变性的可能性。③通过先证者做家系调查时可做为判断是否为症状前或症状早期患者的辅助方法。
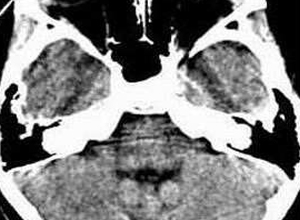
(3)颅脑CT、MRI 无症状的肝豆状核变性及无脑症状的肝型肝豆状核变性患者颅脑CT扫描以脑萎缩为多见,而脑型肝豆状核变性则以基底节区对称性低密度影为特征。因此,CT扫描对不典型的潜伏型、肝型及脑型肝豆状核变性患者都有辅助诊断价值,但肝豆状核变性的CT改变无特异性。肝豆状核变性脑部MRI检查,可显示出比CT更为清晰的颅内异常表现,临床意义与CT扫描相似。侵犯基底节神经核团时均表现为双侧对称性,且为豆状核、尾状核头部的大部分受累,而丘脑则为局部受累。脑干病灶则以脑桥和中脑病变为主,少见小脑病灶。因而,对称性基底节异常信号同时伴有脑干病灶是肝豆状核变性的影像特征之一。
3、电生理检查
(1)脑电图 以脑症状为主的脑型肝豆状核变性患者脑电图多正常或轻度异常,以肝脏损害为主的腹型或肝型肝豆状核变性患者的脑电图多为中度、重度异常。脑电图检查有助于对有癫痫发作的肝豆状核变性进行诊断。
(2)脑干听觉诱发电位(BAEP) 肝豆状核变性患者可出现BAEP异常,有一定的辅助诊断价值。
4、心理测试及IQ检测
对精神障碍型肝豆状核变性或呈现精神症状的其他类型肝豆状核变性,可通过心理测试以区别属于行为障碍或器质性精神病。IQ检测能了解患者智能障碍的程度。
5、其他检查
(1)Tc胶态硫同位素扫描 可清晰地显示肝、脾的大小及形态。
(2)腹腔镜检查 可看到肝脏硬化结节,有助于直接了解肝豆状核变性患者肝脏损害的程度。
诊断:
(1)家族遗传史。父母是近亲婚配、同胞有HLD患者或死于原因不明的肝病者。
(2)缓慢进行性震颤、肌僵直、构语障碍等锥体外系症状、体征及(或)肝症状。
(3)肉眼或裂隙灯证实有K-F环。
(4)血清铜蓝蛋白。
(5)尿铜>50μg/24h。
(6)肝铜>250μg/g(干重)。
判断:凡完全具备上述(1)~(3)项或(2)及(4)项者,可确诊为临床显性型。仅具有上述(3)~(5)项或(3)~(4)项者属无症状型HLD。仅有(1)、(2)项或(1)、(3)项者,应怀疑HLD。
鉴别诊断:
1、Mekes病及慢性肝病由于蛋白严重缺乏,血清CP可下降,胆汁性肝硬化也可出现K-F环,须注意鉴别;
2、本病出现帕金森病某些体征,可根据角膜K-F环、严重共济失调性震颤、血清铜蓝蛋白降低等与PD鉴别;
3、还须与急性或慢性肝炎、肝硬化、小舞蹈病、Huntington舞蹈病、扭转痉挛、老年性痴呆、精神病、肝肾综合征等鉴别。
防治本病应及早确诊,及时纠正患者铜代谢的正平衡状况。注意减少食物含铜量。
对WD患者的家族成员测定血清铜蓝蛋白、血清铜、尿铜及体外培养皮肤成纤维细胞的含铜量,有助于发现WD症状前纯合子及杂合子,发现症状前纯合子可以及早治疗。杂合子应禁忌与杂合子结婚以免其子代发生纯合子。产前检查如发现为纯合子,应终止妊娠,以杜绝患者的来源。