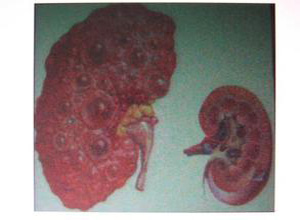
常染色体显性多囊肾即正常肾组织被无数小囊所代替,外形似一串葡萄,有时囊间有岛状正常组织。本病分为4型:常染色体隐性多囊肾;肾发育不良;常染色体显性多囊肾;尿道梗阻多囊肾。其中常染色体显性多囊肾又称成人型多囊肾(ADPKD),发病率约为1/1000,其外显率近乎完全,这使得所有活到80岁以上的携带者均显示出本病的某些征象。约5%~10%终末期肾衰是由常染色体显性多囊肾(ADPKD)导致。
-
挂什么科:内科 肾内科
-
需做检查:肾上腺CT检查 尿常规 肾功能检查 肾脏触诊 肾脏叩诊
-
治疗方法:支持性治疗 手术治疗 康复治疗
-
常用药物:暂无相关信息
-
一般费用:根据不同医院,收费标准不一致,市三甲医院约(10000-50000元)
-
传染性:无传染性
-
治愈周期:如果不进行肾移植,需终身间歇性治疗
-
治愈率:肾移植治愈率约为40%
-
患病比例:本病罕见,发病率约为0.0005%-0.001%
-
好发人群:无特定人群
-
相关症状:
-
相关疾病:
晚期病例肾脏明显肿大可扪及容易诊断,尿液分析见轻度蛋白尿和不同程度的血尿,但红细胞管型不常见,即使并没有细菌感染,脓尿症多见,由于囊肿破裂或结石移动也可有发作性的明显肉眼血尿,静脉尿路造影检查具有特征性,表现为有多个囊肿,及由此引起的肾脏肿大,外形不规则,并且因为囊肿压迫肾盏,漏斗和肾盂而使其拉长呈蜘蛛状,由于囊肿取代功能性组织,故在肝,肾的超声检查和CT扫描中可显示典型的"虫蚀"状,因此在静脉尿路造影未显示典型改变之前,这些检查可作为该病早期诊断的手段,需与该病相鉴别的是尚未造成足够肾实质损害导致尿毒症的单个或多发性囊肿。
正确的遗传学诊断很快可采用,运用重组DNA技术已发现约85%的APD-KD家族中,被称为PKD1的基因突变定位于16号染色体短臂(P)上,它具有两个特异性标志:α球蛋白复合体及磷酸甘油酸激酶的基因,大多数其余的家族发现在4号染色体(PKD2)上有基因缺陷,但也有少数家族不与如何基因座相关。
本症确切病因尚不清楚,尽管大多在成人以后才出现症状,但在胎儿期即开始形成,囊肿起源于肾小管,其液体性质随起源部位不同而不同,起源于近端小管,囊肿液内成分如Na+,K+,CI-,H+,肌酐,尿素等与血浆内相似;起源于远端则囊液内Na+,CI-浓度较低,而K+,H+,肌酐,尿素等浓度较高。
染色体异常(35%):大多数患者的异常基因位于16号染色体的短臂,称为ADPKD1基因,基因产物尚不清楚,少数患者的异常基因位于4号染色体的短臂,称为ADPKD2基因,其编码产物也不清楚,两组在起病,高血压出现以及进入肾功能衰竭期的年龄有所不同。
诊断
根据病史,临床表现及实验室资料不难做出诊断。
鉴别诊断
主要考虑与多房性单纯肾囊肿鉴别,其他鉴别需考虑ARPKD,获得性肾囊肿,多囊性肾发育不良等,表19-30早期ADPKD与多房性单纯性肾囊肿鉴别。
多房性肾囊肿(multilocular cyst of kidney)是指肾内有局限性、大而有完整被膜的肿物,压迫周围肾组织,内由多个囊肿构成。
常染色体显性多囊肾西医治疗
约50%的具有ADPKD1突变的患者在55~60岁之间发展到尿毒症。而非ADPKD1突变的要到70岁才发生。少数ADPKD患者在少儿时就出现临床表现,即使其父母成年后发病。与许多其他肾脏疾病一样,ADPKD在黑人中有进展加速,约提早10年发病。其他预示该病更快进展的因素包括年幼时即诊断,男性,肾脏体积较大,高血压,肝囊肿(在女性患者中),肉眼血尿及尿路感染(男性)。如未进行透析或肾移植,患者常死于尿毒症或高血压并发症,约10%的患者死于动脉瘤破裂引起的颅内出血。对尿路感染和继发性高血压进行有效的治疗可明显延长生命。若尿毒症存在其处理与其他肾脏病相同。ADPKD透析治疗患者的血红蛋白水平高于其他类型的患者。肾移植是可行的,但由于该病的家族特性,使用双亲和同胞供肾脏。
常染色体显性多囊肾中医治疗
当前疾病暂无相关疗法。 (仅供参考,详细请询问医生)
一、保持心情舒畅和乐观向上的情绪,树立起战胜疾病的信心。囊肿性疾病是先天和后天各种因素相互作用的结果,科学研究发现,所有这些因素都是可以改变或加以控制消除的,因此,千万不可悲观失望,况且乐观向上的思想情绪可以提高人的免疫力,有利于战胜疾病。但另一方面也要克服“轻敌”思想,积极地配合医生进行治疗。乐观向上,认真对待才是正确的指导思想。
二、及时治疗,科学用药:多囊肾是一种遗传性疾病多伴有多囊肝,50%并发高血压。它们的生成不断压迫肾脏组织,致使肾脏血液循环受阻。
与患者年龄,起病年龄,高血压的控制程度,是否反复发作尿路感染,血尿等有关,随着透析及肾移植技术的不断提高,患者的主要死因为感染,心血管疾患(心梗,心衰等)以及颅内出血。
影像学检查,包括超声波,CT及磁共振等。
CT是用X射线束对人体某部一定厚度的层面进行扫描,由探测器接收透过该层面的X射线,转变为可见光后,由光电转换变为电信号,再经模拟/数字转换器(analog/digital converter)转为数字,输入计算机处理。图像形成的处理有如对选定层面分成若干个体积相同的长方体,称之为体素(voxel)。
尿路感染最常见,大多为下尿路感染,也可出现肾盂肾炎,囊肿感染等,其他并发症有:尿路结石,梗阻;动脉瘤破裂出血,特别是颅内动脉瘤破裂占ADPKD患者死因的7%~13%,极少见情况下可出现两肾的恶性肿瘤。