黏脂贮积症Ⅱ型又称包涵体细胞病(inclusion cell disease),简称I-cell病。其临床特征更像Hurler综合征,表现为出生时就有明显的临床和X线异常,反应迟钝,但无黏多糖尿症,皮肤成纤维细胞培养有大量粗大细胞质包涵体,Leroy等(1967)最早发现此病,Spranger(1970)将其分类为黏脂贮积症Ⅱ型。
出生时即可发现许多异常,如先天性髋关节脱位,男性婴儿腹股沟疝,面容粗笨,骨骼异常,运动受限和全身性肌张力低下等,但在新生儿期一般不能做出诊断,大约在出生后6个月,婴儿的身长可在正常范围内,但有全身性肌张力低,不能在床上滚动,头支撑不良和许多外观异常,部分病例可有重度智力低下。
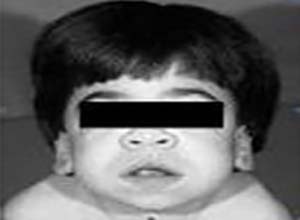
面容呈进行性粗笨,前额高,内眦有赘皮,眼睑肥厚,鼻梁扁平,鼻口上翻,齿龈增生,角膜清晰,但经裂隙灯检查可发现有细微的弥漫性基质异常,患儿鼻涕增加,可反复发生呼吸道感染,肺炎和中耳炎,关节运动受限,并有挛缩,皮肤紧而增厚,腹部膨隆,肝脏增大,运动迟钝,一般在一岁以后可出现心脏收缩期杂音,短颈,胸廓畸形及小头畸形等。
(一)发病原因
黏脂贮积症的病因是常染色体隐性遗传。
(二)发病机制
本症是由多种水解酶缺陷引起的,利用病人成纤维细胞混合培养发现,此症的基本生化缺陷是几种溶酶体酶的识别部位异常,溶酶体酶的正常组合需要细胞内的协同作用,即细胞合成,分泌水解酶,细胞表面识别水解酶,继之被溶酶体摄取,并固定于溶酶体内,细胞表面识别水解酶,需要在细胞表面和溶酶体水解酶上都有特异性部位,目前认为黏脂贮积症Ⅱ型系基因突变引起的几种溶酶体酶发生识别部位缺损,也就是酶分子结构中的识别亚基异常所致。
Strecker等(1976)发现此症病人组织细胞神经氨酸酶缺乏,可能该种酶缺乏与多种溶酶体水解酶的识别部位异常有关,如己糖苷酶,β-葡萄糖苷酸酶,β-半乳糖苷酸酶等的识别部位缺损,造成过多的中性,酸性黏多糖及黏脂沉积于组织细胞内而致病。
病理:组织学改变主要限于间质细胞,肝组织活检显示大多数库普弗细胞形态正常,而肝细胞内充有各种类型的包涵体,可含有无包膜的脂肪小滴,也有0.7~50µm的包涵体,这些包涵体表现为不同程度的长方形结晶,并位于微丝物质内,微丝物质外包有一层膜,肝细胞内最多的一种包涵体含有亲水物质,该物质具有成层结构或呈小球状,肾脏的包(Bowan)氏囊细胞显示有类似肝细胞所存有的包涵体,其大小为0.3~4.0µm,PAS染色呈阳性,脑组织的神经元和胶质细胞在显微镜下显示正常,但在电子显微镜下可见神经元,星状细胞和血管周围上皮细胞有0.3~1.5µm的透明包涵体,周围有包膜空泡,空泡内有均匀而细小的颗粒基质,这种基质含有少量的成层结构,还有较致密的,周围有包膜的亲水包涵体,髂骨嵴活检显示软骨细胞质内含有大量溶酶体,溶酶体内含有细小,网状,颗粒状及有膜的包涵体,组织化学染色显示包涵体内沉积有中性,酸性黏多糖和黏脂,显微镜检查可发现软骨内骨化明显障碍,伴有完全性增生,肥大性软骨脱钙区,原有的骨小梁短,近骨干处混有持续存在的软骨区,所有器官的成纤维细胞内均含有大量周围有包膜的空泡,从而使成纤维细胞呈明显的气球样,周围淋巴细胞也可有多数大空泡,所有器官成纤维细胞内的包涵体均为多形性,从透明空泡到致密亲水小体或成层排列不等。
根据临床症状,X线表现,实验室检查所见以及尿中无过多黏多糖排出,可初步诊断为本病,培养的皮肤成纤维细胞及各种组织内有包涵体,溶酶体内有多种酶缺 陷,如β-半乳糖苷酸酶,N-乙酰-半乳糖胺酶,β-葡萄糖胺酶,芳基硫酯酶,岩藻糖苷酶等,而血清中这些酶的活性增高,电镜检查可见溶酶体肿胀,其内充 以有包膜的致密物质,即可诊断。
本型黏脂贮积症需与其他型黏脂贮积症相鉴别。
黏脂贮积症Ⅱ型西医治疗
无特殊疗法。畸形严重者应予手术矫形。对感染和心力衰竭的病人,应进行对症处理。
预后
预后不良,通常在2~8岁内死亡,多死于感染和心力衰竭。
黏脂贮积症Ⅱ型中医治疗
当前疾病暂无相关疗法。
(以上提供资料及其内容仅供参考,详细需要咨询医生。)
饮食控制(禁其所忌):当代谢异常造成机体某些必需物质缺乏时,通过饮食加以补充;而当代谢物质发生贮积时,则限制此代谢物或其前身物质的摄入,来维持平衡。苯酮尿症患者低苯丙氨酸饮食就是很好的范例。另外,还可通过限制对特定物质的吸收来减少摄入,如苯酮尿症患者服用苯丙氨酸氨基水解酶胶囊,可以将食物中的苯丙氨酸转化为转苯丙烯酸,而被消除。
1.一级预防
遗传病的预防,除了从整个人群的角度做好流行病学调查,携带者检出,进行人群遗传监护和环境监护,开展婚姻和生育指导,努力降低人群中遗传病发生率,提高人口素质之外,针对个体,必须采取有效的预防措施,避免遗传病后代的出生(即实行优生)和遗传变异的发生,采取通常的措施包括:婚前检查,遗传咨询,产前检查和遗传病的早期治疗。
(1)婚前检查:婚前检查(即婚姻保健),它是保证男女双方婚后生活幸福,后代健康的重要环节,婚前检查的重点是:
①遗传病方面的调查,包括详细询问男女双方及其家庭成员的健康状况,既往病史及医治情况,尤其是有无先天畸形,遗传病史和近亲婚配史,必要时应进行家系调查,血型检查,染色体检查或基因诊断,以检出携带者;
②全面的体格检查,主要是对急性传染病,结核病,或严重的心,肝,肾疾病,泌尿道慢性炎症等可严重威胁个人或配偶健康的疾病,以及女方的严重贫血,糖尿病等可对胎儿造成影响的疾病的检出,并动员经治愈后才可结婚;
③对男女生殖器官的检查,检出性器官畸形,两性畸形等疾患,以便极早采取措施。
(2)遗传咨询:遗传咨询(genetic counselling)是由临床医生和遗传学上,作肯定解答,遗传病患者及其亲属提出的有关遗传性疾病的病因,遗传方式,诊断,治疗及预后等问题,估计患者的子女再患某病的概率,并提出建议及指导,以供患者及其亲属参考,遗传咨询的意义在于:
①减轻患者身体和精神上的痛苦,减轻患者及其亲属的心理压力,帮助他们正确对待遗传病,了解发病概率,采取正确的预防,治疗措施;
②降低人群遗传病的发生率,降低有害基因的频率,及减少传递机会。
2.遗传病治疗中总的原则是禁其所忌,去其所余,补其所缺,调节代谢平衡,防止症状的出现。
(1)纠正代谢紊乱:这是目前治疗遗传性代谢病的最主要方法,随着对遗传性代谢病发病机制和中间过程的认识不断深化,此法的适用范围也日益扩大。
①饮食控制(禁其所忌):当代谢异常造成机体某些必需物质缺乏时,通过饮食加以补充;而当代谢物质发生贮积时,则限制此代谢物或其前身物质的摄入,来维持平衡,苯酮尿症患者低苯丙氨酸饮食就是很好的范例,另外,还可通过限制对特定物质的吸收来减少摄入,如苯酮尿症患者服用苯丙氨酸氨基水解酶胶囊,可以将食物中的苯丙氨酸转化为转苯丙烯酸,而被消除。
②减少底物(去其所余):因代谢产生有害物质而引起疾病时,可以通过降低有害底物和减少其前身物质及代谢衍生物的浓度,去除或减少其毒性作用来控制或改善疾病的症状,主要方法有:A.螯合或促进排泄;B.血浆置换法和亲和结合法;C.改变代谢途径;D.外科旁路手术;E.代谢抑制。
③产物替代(补其所缺):当重要的酶促反应产物不足而致病时,可直接补充相应的必需的终产物,如给垂体性侏儒患者以生长激素,给血友病患者以抗血友病蛋白(凝血因子),给遗传性免疫缺陷病人以相应的免疫球蛋白。
(2)纠正酶活性异常:
①辅酶的补充:有些遗传病,酶活性异常可能累及:
A.一种特异性辅酶或维生素的结合部位。
B.有活性的辅酶转运或生物合成过程,导致异常,许多辅酶是全酶正常活性所必需的,所以补充辅酶成分也是诱导酶活性增加的一种有效方法,它可以使全酶在细胞内降解速度减慢,提高酶的半衰期,还可降低酶促反应的米氏常数(Km),目前已用此方法治疗25种以上的遗传病,如用钴胺素(B12)治疗多种贫血和甲基丙二酸尿症等。
②酶诱导或反馈抑制:对酶缺陷水平的另一种疗法是用药物来提高残余酶活性以改善代谢水平,例如苯巴比妥和有关药物能明显刺激滑面内质网的生成,并能加速内质网中特异性酶合成,包括肝UDP葡萄糖醛酸转移酶,为用苯巴比妥治疗Gibert综合征和Crigler-Najjar综合征提供了理论基础。
反馈抑制作用是许多代谢调节中的重要形式,针对因某种酶缺陷引起的底物或其前体堆积,可以通过其他旁路代谢的反馈抑制作用来提高酶活性,减少堆积的底物,反馈抑制已作为治疗急性卟啉症的一种方法。
③同种移植:通过向遗传病个体植入同种含正常基因的细胞,组织或器官,以期在受体内产生相应的有活性的酶及其他基因产物,达到治疗目的,移植物在受体内可能通过两种机制发挥作用:
A.产生活性酶,在原位代谢除去原来的贮积底物。
B.释放活性酶,辅酶或免疫活性因子入血,分布到全身其他组织中发挥作用,至今已进行过此类同种移植的组织器官有:肾,肝,肾上腺,骨髓,胸腺,脾,胰等,有的已取得明显疗效。
④酶替代疗法:直接给酶缺陷患者提供相应的正常的酶,随着酶学技术和细胞工程,基因工程技术的发展,已经可以提供足量的,高纯度的酶制剂,这种酶制剂必须具有半衰期长,抗原性低,导向性好等特性,为此常采用的方法是:
A.采用微囊,脂质体,红细胞影泡等载体来包装酶制剂,以减小免疫原性,延长半衰期。
B.应用受体介导分子识别法来提高导向性。
C.对一些溶酶体贮积病,因其沉积物可以弥散入血,并保持动态平衡,则可用“平衡-去除”法来治疗。
(3)基因治疗:基因治疗是指运用基因转移技术直接将遗传物质导入生殖细胞或体细胞以起到对遗传病及其他疾病的治疗作用的新型治疗方法,对遗传病进行基因治疗可望从根本上纠正遗传病的表型异常。
①基因治疗的基本策略:近十余年来,基因治疗研究蒸蒸日上,提出了许多新思路,新设想,目前主要的策略有:
A.基因的原位修正(correction)和原位替代(replacement),这一策略的目的就是要将突变的基因在原位修复,而不影响其周围其他基因的结构和功能,其中原位修正针对基因的点突变或小范围变异,拟通过特定方法对其定点修复,而原位替代,就想把有较大范围变异的基因去除而换之以正常的基因,这一策略是最理想,最直接的对遗传变异进行根治的方法,目前研究很多的哺乳动物细胞内定点整合(同源重组),给这种策略提供了理论和实验依据,但至今未能真正用于人体试验。
B.基因增强(gene augmentation或gene complementation),在不改变缺陷基因本身的前提下,将外源有功能的基因转移到疾病细胞或个体基因组内,使其表达以补偿有病基因失去的功能,此策略是目前研究最多,也是最成熟的方法。
C.将反义基因或其他对抗异常基因表达产物的基因导入细胞内,起到抑制作用,或称基因抑制疗法(gene inhibition therapy)或细胞内免疫(intercellular immunity)。
②基因治疗的技术要点在基因治疗的诸多策略中研究最多,最成熟并应用于临床试验的是基因增强的策略,整个研究过程通常包括临床前研究和临床研究。
A.疾病的选择:目前基因治疗首选的是单基因缺陷性疾病,选择的基本条件常包括:
a.遗传基础比较明确,目的基因能在体外克隆。
b.基因表达不需精细调节,而且经常开放,产物生理水平不高者更佳。
c.具有一定发病率,危害较大,尚缺乏其他有效治疗措施者。
我国是开展基因治疗研究较早的国家之一,复旦大学薛京伦等就是根据这些条件,选择血友病作为研究对象,已取得了很好的结果,达到了世界先进水平,当然,这些条件是限于现有的研究水平才提出的。
B.靶细胞的选择:基因治疗的靶细胞可分为两大类:生殖细胞和体细胞,由此引出了生殖细胞基因治疗和体细胞基因治疗的分类,如果能对生殖细胞或早期胚胎细胞进行基因修复或替换,使基因缺陷得到校正,使遗传病不但能在当代得到治疗,还能将新基因传给下一代,也为人群减少一个有害基因,是理想的遗传病根治手段,但是,由于现代生物技术,理论的限制,以及生殖细胞基因操作涉及人类社会的伦理,道德和法律等多种因素,在相当长的一段时间内只能进行动物试验,1985年美国政府就已规定,把基因治疗的人体试验限制在体细胞,已经被用于作为靶细胞的有:造血干细胞,肝细胞,成纤维细胞,内皮细胞,淋巴细胞等。
C.基因转移的载体和转移方法:构建合适的转移并表达的载体和选择高效的基因转移方法是基因治疗的关键,常用的载体有:逆病毒载体,质粒载体和腺病毒载体,腺相关病毒载体,另外还有脂质体载体,常用的基因转移方法有四大类型:
a.化学法:主要是磷酸钙沉淀法。
b.物理法:常用电导和显微注射法。
c.膜融合法:以脂质体包裹法较好。
d.病毒法:主要指反转录病毒和腺病毒介导的基因转移。
③基因治疗的前景:基因治疗概念的提出已有几十年的历史,只到了近10年,随着现代分子生物学技术(特别是DNA重组技术)的发展,这一概念才得到有力的理论基础和技术方法的支持,并得以付诸实施,1990年,两名腺苷脱氨酶(ADA)缺陷引起严重免疫缺陷的患者接受基因治疗获得成功,这标志着基因治疗的研究进入了一个新的阶段,从此世界各国的生物医学家,在各国政府部门及社会各种力量的大力支持下,全面展开了基因治疗的研究,由原来针对单一的遗传病发展到肿瘤,传染病等多种疾病,提出了基因调控疗法,基因抑制疗法等新概念,新途径,到1994年上半年,已有100多个临床试验方案获准实施,有的已取得很好的效果,当然,基因治疗发展的历史还不长,要广泛应用于临床还需大量的研究探索,尤其是以下几方面的问题:
A.对更多遗传病的分子基础及基因表达调控机制更深入的了解,这是基因治疗的基础。
B.构建更有效和安全地表达并转移的载体。
C.更简便有效的基因转移方法的建立。
D.定点整合,原位修复系统等技术的完善。
E.更多更接近实际的动物模型(尤其是转基因动物模型)的建立,这是基因治疗临床前试验的必由之路。
F.体细胞基因治疗,生殖细胞基因治疗等的伦理学及相关的科技管理立法等方面的探讨。
G.还需充分考虑基因治疗可能存在的危害性,如插入突变导致的严重后果,缺陷病毒载体经重组后恢复感染性的危害及外源基因转入体内的其他潜在危害等,总之,我们认为基因治疗作为一种惟一从基因缺陷本身入手,可望彻底治疗遗传病新型治疗途径,有非常吸引人的前途,但仍需从基础理论,技术方法及伦理道德等多方面进行深入广泛的研究探索,才能适应现代医学模式,被人们所接受,真正成为人类防病治病的有效手段。
尿中无过多的酸性黏多糖排出,培养的皮肤成纤维细胞及各种组织内有包涵体,溶酶体内有多种酶缺陷,如β-半乳糖苷酸酶,N-乙酰-半乳糖胺酶,β-葡萄糖胺酶,芳基硫酯酶,岩藻糖苷酶等,而血清中这些酶的活性增高,电镜检查可见溶酶体肿胀,其内充以有包膜的致密物质。
X线检查:X线表现为多发性骨发育不良,早期幼儿有明显的骨膜新骨形成,这种改变尤以股骨和肱骨的骨干周围为显著,在两岁以内,骨发育不良可呈进行性加重,到两岁时,上肢长管骨粗短,髂骨发育不全,髋臼变浅,脊柱改变以下胸椎和上腰椎处为著,椎突短而圆,呈鸟嘴状,可发生明显的胸腰部驼背,肋骨呈船桨状,掌骨不规整,增宽,呈圆锥样,指骨呈子弹状,颅骨增厚,可有心脏扩大及肺部感染征象。
婴儿可并发先天性髋关节脱位,男性婴儿可并发腹股沟疝,还可并发肝脏增大,一岁以后可出现心脏收缩期杂音,短颈,胸廓畸形及小头畸形等。